以下は、Qiime2公式ドキュメント(1)に基づき、qiime dada2 denoise-paired コマンドの主なパラメータについて日本語で解説した学習ノートです。Qiime2でペアエンドリードを処理する際の参考になれば幸いです。
DADA2処理の流れ(概略)
Truncate&Trim → Learn error → Denoise → Merge → Chimera removal → ASV table生成
入力ファイル
-
--i-demultiplexed-seqs
入力ファイル(demultiplexed SampleData[PairedEndSequencesWithQuality])
シーケンスのトリミングと除去
-
--p-trunc-len-f / --p-trunc-len-r
3' 末端をトリミング(シーケンス後半の質が落ちる部分を除去)。
→ リードの右端をカット
→ 設定値より短いリードは破棄される
→ ForwardとReverseのマージには、少なくとも12塩基以上のオーバーラップが必要(--p-min-overlap) -
--p-trim-left-f / --p-trim-left-r
5' 末端をトリミング(シーケンス前半の質が低い部分を除去)。
→ プライマー配列除去にも利用可能
エラーモデルの学習
-
--p-n-reads-learn
エラーモデル学習に使用するリード数(デフォルト:1,000,000)
品質フィルタリング
-
--p-max-ee-f / --p-max-ee-r
期待エラー数(Expected Errors)がこの値を超えるリードを破棄
→ デフォルトは2.0
DADA2の最初の切り捨て作業(Filter&Trim)の後に、一定のリード(Qiime2のデフォルトでは100万)を抽出して、エラーモデルの学習を行います。得られたエラーモデルはQiime2上では確認できませんが、Rで動かすと以下のようなグラフを得られます:
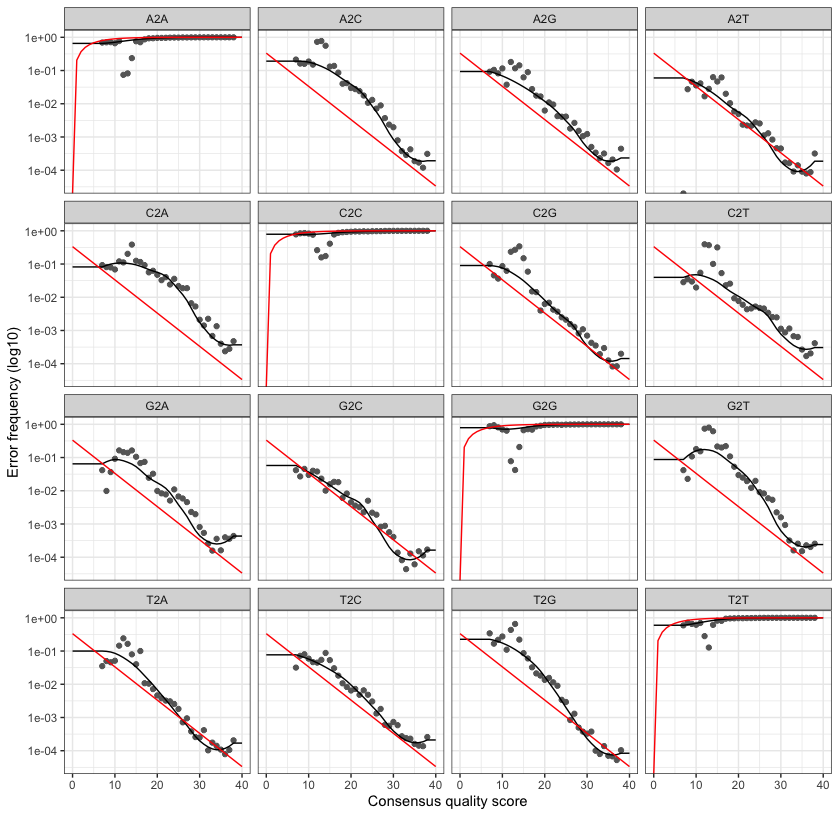
このモデルは、各Quality値ごとに A→C、A→G、A→T などの読み間違いの頻度を推定するものです。ドットは実際に観測されたエラー率、赤線はQスコアから推定された期待値です。
そこから各リードの合計 expected error(EE)=各塩基の期待エラーの合計が計算され、設定した --p-max-ee よりも高い場合、そのリードは破棄されます。
📎 補足:エラーモデルはランごとに作成する
シーケンサーのランごとにエラーの傾向が異なるため、Qiime2公式ドキュメントでは「**各シーケンスランごとに**DADA2による denoise を実行し、その後マージする」ことが推奨されています。The DADA2 denoising process is only applicable to a single sequencing run at a time, so we need to run this on a per sequencing run basis and then merge the results. We’ll work through this initial step, and then pose several questions that can be answered as an exercise.
-
--p-trunc-q
Quality score が指定値以下になった時点でリードをカット
→ カットされたリードが 最初の--p-trunc-lenより短いと破棄される
オーバーラップとマージ
-
--p-min-overlap
Forward/Reverse のマージに必要な最小オーバーラップ長
→ デフォルトは12
サンプルの処理方法
-
--p-pooling-method(Choices: 'independent', 'pseudo')
'independent': サンプルごとに独立にDenoise(デフォルト)
'pseudo': 2回Denoiseを行い、2サンプル以上で現れたASVに対する感度を上げる
💡 Rでの設定例
R環境では `dada(..., pool="pseudo")` と指定します。キメラ除去
-
--p-chimera-method(Choices: 'consensus', 'none', 'pooled')
'consensus': サンプル単位で検出し、一定割合以上で除去(デフォルト)
'pooled': 全サンプルをまとめて検出
'none': キメラ除去を行わない -
--p-allow-one-off
1つのミスマッチやindelを許容するキメラ(Bimera)も検出
True: 厳しめの検出。1塩基のミスマッチやインデルを許容するキメラ(one-off bimera)も対象
False: 厳密な一致があるキメラ(exact bimera)のみ対象(デフォルト)
その他のパラメータ
-
--p-hashed-feature-ids
Trueにすると、ASV IDが配列のハッシュ値になる。異なるランの統合解析に有用(デフォルト:True) -
--p-retain-all-samples
True:全サンプルを保持
False:ASV頻度が0のサンプルを除去
出力ファイル(3点)
-
--o-table:FeatureTable[Frequency](各サンプル×ASVの頻度テーブル) -
--o-representative-sequences:FeatureData[Sequence](各ASVの代表配列(Qiime2上でクリックするとBLAST検索が可能) -
--o-denoising-stats:SampleData[DADA2Stats](各ステップでのリード数の推移)
使用例
qiime dada2 denoise-paired \
--i-demultiplexed-seqs input.qza \
--p-trunc-len-f 280 \
--p-trunc-len-r 180 \
--p-trim-left-f 22 \
--p-trim-left-r 23 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats stats-dada2.qza