1pwcはβ-lactamaseのSer側鎖とpenicillin Gとが共有結合した複合体です。本記事ではAutoDock4 (ad) のcovalent dockingで、β-lactamaseのapo体から1pwcを再現します。
adでcovalent dockingを行うにはMGLToolsに含まれていないpythonスクリプトを使用する必要があります。そこでCovalent Docking - AutoDockからadCovalentDockResidue_v1.2.tar.gzをダウンロードし、コマンドプロンプトで解凍します。本記事は、解凍後のフォルダのREADME.txtに沿ってcovalent dockingします。
tar -xvf adCovalentDockResidue_v1.2.tar.gz
1. ファイルの準備
ドッキングに必要なligandとreceptorとを準備します。ligandのpenicillin GはPubChemから3次元構造をダウンロードしてきます。またreceptorのβ-lactamaseはPDBからダウンロードしてきます。
1-1. receptorの準備
PDBからβ-lactamaseのapo体である3PTEをダウンロードします。PyMOL等で3pte.pdbから結晶水を消去し、receptor.pdbとして保存します。
1-2. ligandの準備
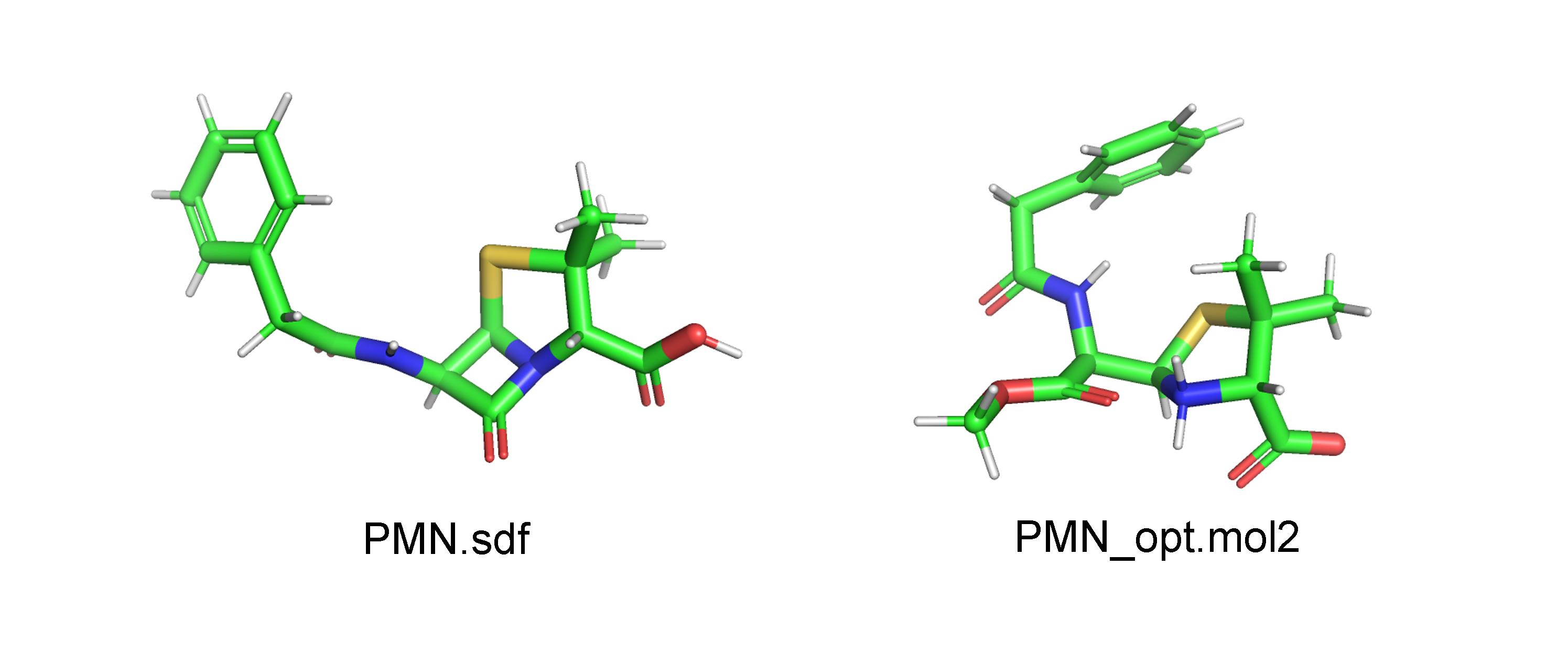
結合する残基がSerである場合、構造にMeOHを付加したligandを作成します。初めにPubChemから3次元構造(sdf形式)をダウンロードし、ファイル名をPNM.sdfに変更します。続いてPyMOL等でMeOHが付加した開環体をモデリングし、PNM_MeOH.mol2(mol2形式)として保存します。ダウンロードしてきた3次元構造ではプロトン化状態が中性付近のものでないため、その修正もここでしておきます。最後にPNM_MeOH.mol2をChem3D等でエネルギー最小化し、PNM_MeOH_opt.mol2として保存します(図1)。
図1. PNM.sdfとPNM_MeOH_opt.mol2
2. prepareCovalent.py
先ほどダウンロードしたpythonスクリプト(prepareCovalent.py)で、receptorのSer側鎖とligandに付加したMeOHと重ね合わせます。
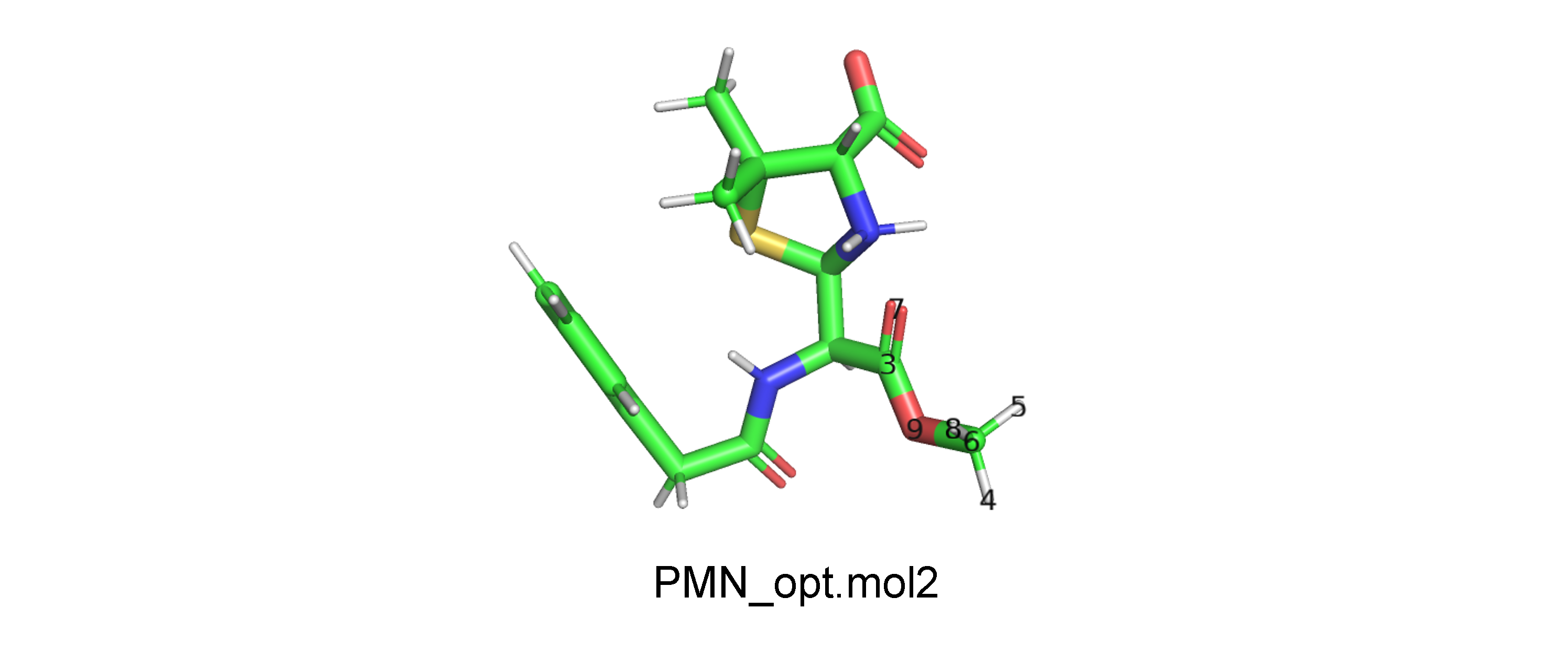
まず、PNM_MeOH_opt.mol2においてMeOHのC, Oが何番目の原子であるのかを確認します(図2)。Cが6番目、Oが9番目であることから、--ligindicesを6,9とします。
図2. PNM_MeOH_opt.mol2のMeOH部分
続いて、receptor.pdbにおいてどのSer残基がligandと共有結合するかを確認します。先行研究からreceptor.pdbはChainA 62番目のSer側鎖でpenicillin Gと結合していることから、--residueをA:SER62とします。
mgltoolsをインストールした仮想環境で以下を実行するとligcovalent.pdbが出力されます。
pythonsh /path/to/adCovalentDockResidue/adcovalent/prepareCovalent.py \
--ligand PMN_MeOH_opt.mol2 \
--ligindices 6,9 \
--receptor receptor.pdb \
--residue A:SER62 \
--outputfile ligcovalent.pdb
receptor.pdbとligcovalent.pdbとを描画すると、receptorとligandとが衝突していることがわかりますが、ここでは問題ありません(図3)。
図3. receptor.pdbとligcovalent.pdbとの衝突
3. PDBQTへの変換
receptor.pdbとligcovalent.pdbとをadで取り扱うためPDBQT形式に変換します。mgltoolsをインストールした仮想環境で以下を実行すると、それぞれをpdbqt形式に変換したファイルが出力されます。
# receptor.pdbの変換 (receptor.pdbqtが出力される)
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_receptor4.py \
-r receptor.pdb \
-o receptor.pdbqt \
-U nphs_lps_waters \
-v
# ligcovalent.pdbの変換 (ligcovalent.pdbqtが出力される)
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_ligand4.py \
-l ligcovalent.pdb \
-o ligcovalent.pdbqt \
-v
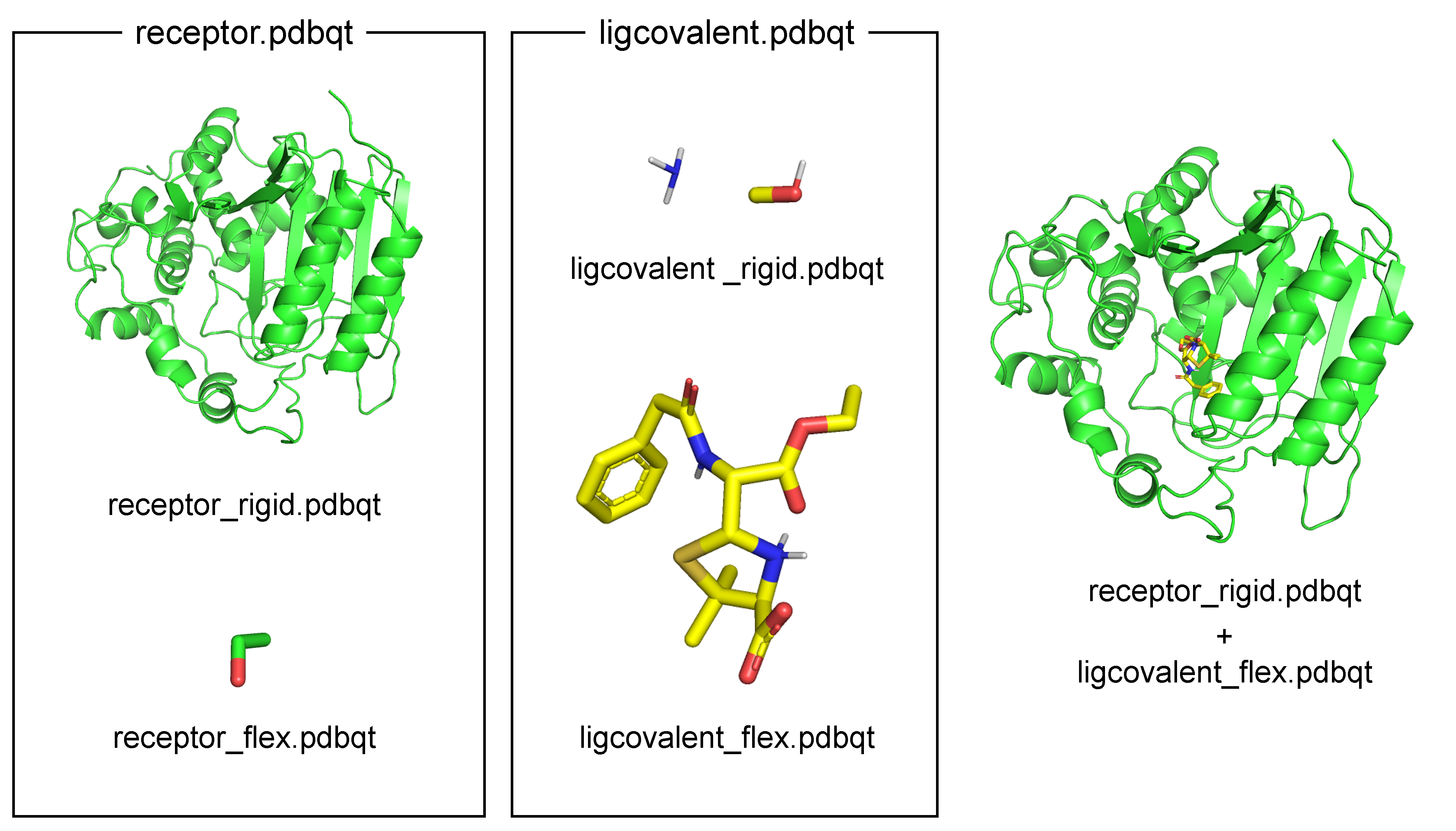
4. flexible/rigid PDBQTの生成
adのCovalent Dockingではligandをflexible residueとして扱います。そのためにreceptor.pdbqtとligcovalent.pdbqtとを、それぞれflexible partとrigid partとに分けます。
# receptor.pdbqtの変換 (receptor_rigid.pdbqt、receptor_flex.pdbqtが出力される)
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_flexreceptor4.py \
-r receptor.pdbqt \
-s receptor:A:SER62
# ligcovalent.pdbqtの変換 (ligcovalent_rigid.pdbqt、ligcovalent_flex.pdbqtが出力される)
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_flexreceptor4.py \
-r ligcovalent.pdbqt \
-s ligcovalent:A:SER62
Covalent dockingにはreceptor.pdbqtのrigid partとligcovalent.pdbqtのflexible partとを使用します(図4)。
図4. AutoDock4のCovalent dockingで使用するファイル
5. パラメータファイルの生成
ドッキング条件を指定するパラメータファイルを出力します。以下を実行すると、
receptor.gpfとligcovalent_receptor.dpfが出力されます。
# .gpf (AutoGrid用のパラメーターファイル) の生成
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_gpf4.py \
-r receptor_rigid.pdbqt\
-x ligcovalent_flex.pdbqt\
-l ligcovalent_flex.pdbqt\
-y -I 20\
-o receptor.gpf
# .dpf (AutoDock用のパラメーターファイル) の生成
pythonsh /path/to/envs/mgltools/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_dpf4.py \
-r receptor_rigid.pdbqt\
-x ligcovalent_flex.pdbqt\
-l ligcovalent_flex.pdbqt\
-o ligcovalent_receptor.dpf\
-p move='empty'
# 空のファイルを作成しておく。
touch empty
生成されたligcovalent_receptor.dpfをメモ帳等で開き、unbound_model boundをunbound_energy 0.0に書き換えます。さらにga_runの値を10から100に書き換え、出力されるドッキングポーズを100個にします。
6. ドッキング
最後にドッキングします。autogrid4とautodock4とをインストールした仮想環境で以下を実行し正常に終了すると、ドッキングモデルが記載されたligcovalent_receptor.dlgが出力されます。
autogrid4 -p receptor.gpf -l receptor.glg
autodock4 -p ligcovalent_receptor.dpf -l ligcovalent_receptor.dlg
結果はadtから確認します。mgltoolsをインストールした仮想環境でコマンドプロンプトにadtと入力し実行すると、AutoDockToolsの画面が現れます。
- 画面右下の”Remind Me Later”が赤くなり次第、クリックする
-
Analyze → Docking → Open...によりligcovalent_receptor.dlgを開き、ドッキングポーズを表示する -
Analyze → Macromolecule → Open...によりreceptor_rigid.pdbqtを開き、receptorを表示する -
Analyze → Conformations → Play, ranked by energy...によりドッキングポーズをエネルギーが低い順にソートする - 一つずつドッキングポーズを確認し、確からしいポーズを選択する
-
右から2番目のボタン → Write Complexから表示しているポーズを保存する(図5)。
図5. ドッキングポーズを保存するときの操作

7. 答え合わせ
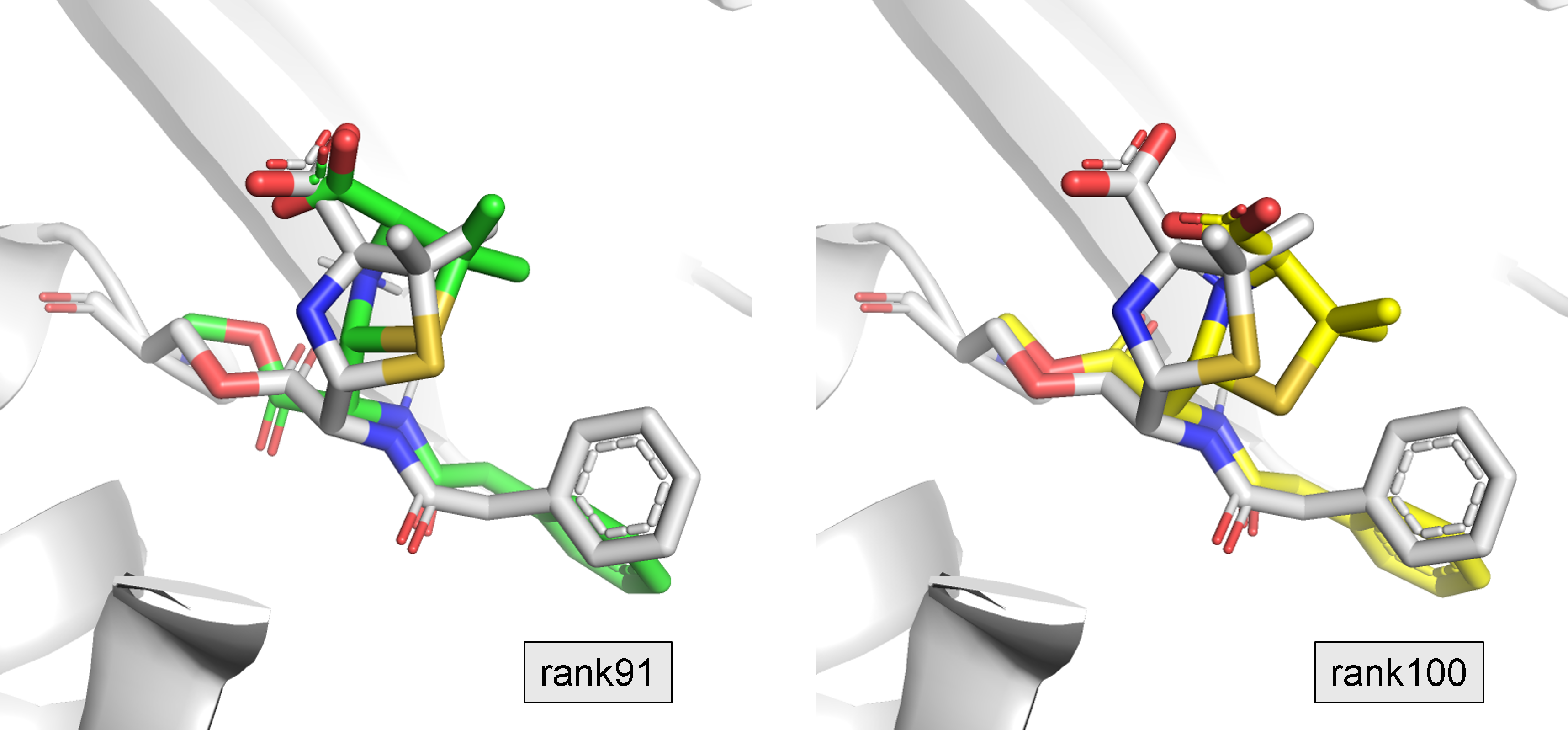
保存したドッキングポーズとX線結晶構造とを比較します。それらをPyMOLのalign機能で重ね合わせて答え合わせします。今回のドッキングシミュレーションでは出力した100ポーズの内、rank91とrank100とがX線結晶構造に近いということがわかりました。しかしrank91ではエステル結合の向きが、rank100ではカルボン酸の位置が1pwcと異なっています。また今回のように、設定条件にも依りますが、正しいポーズのランクが必ずしも良いとは限りません。
図6. rank91、rank100、X線結晶構造の重ね合わせ(灰色:1pwc、緑色:rank91、黄色:rank100)