概要
タンパク質ドッキングとは,2つのタンパク質の構造情報をもとに複合体の構造を推定することです.
この記事ではMEGADOCKというツールを使ってタンパク質ドッキングをやってみるところまで解説します.
インストール
必要なもの
今回は普通のPCで動かす場合の説明をします.
GPUとかスパコンとか使いたい人はhttp://www.bi.cs.titech.ac.jp/megadock/install_ja.html を参照して下さい.
- MEGADOCK
- FFTW3 - http://www.fftw.org
FFTW3のインストール
FFTWのサイト http://www.fftw.org からFFTW3系のtarballを取ってきます.その後,以下のようにインストールして下さい.
$ tar xzf fftw-3.X.X.tar.gz
$ cd fftw-3.X.X
$ ./configure --enable-float --enable-sse2 --prefix=/as/you/like
(SIMD化が効く場合,--enable-sse または --enable-sse2 オプションによってSSEまたはSSE2を有効にすると性能向上が見込めます.)
$ make
$ make check
$ make install
MEGADOCKのインストール
MEGADOCKのサイト http://www.bi.cs.titech.ac.jp/megadock/ からmegadock-4.0.tgzをダウンロードします.その後以下の操作で解凍します.
$ tar xzf megadock-4.0.tgz
$ cd megadock-4.0
次にMakefileを編集します.Makefileの該当箇所を以下に沿って編集して下さい.(FFTWのパスはインストール時に設定したものを入力して下さい.)
FFTW_INSTALL_PATH ?= your/fftw/library/install/path
(デフォルト: /usr/local )
CPPCOMPILER ?= icpc, g++ など
OPTIMIZATION ?= -O3
OMPFLAG ?= -openmp (intel) または -fopenmp (g++)
USE_GPU := 0
USE_MPI := 0
そしてmakeします.
$ make
すると,megadockという名前のバイナリファイルが生成されます.
MEGADOCKを使う
MEGADOCKに2つのPDBファイルを入力すると,ドッキング計算をしてくれます.
$ ./megadock -R A.pdb -L B.pdb -o docking.out
-o [filename] : 出力ファイルの名前 (デフォルト:"$R-$L.out")
-O : 詳細な出力ファイルを生成します
-N [integer] : 生成する候補構造の数 (デフォルト:2000)
-t [integer] : リガンド回転角ごとに出力する候補構造の数 (デフォルト:1)
-D [integer] : リガンド回転角の刻み幅を6°刻みにします(-Dを付けない場合は15°刻み)
6°刻みの場合54000通りの角度,15°刻みの場合3600通りの角度で計算します.
(例えば -t 2 -D とした場合,-Nは最大108000まで設定可能です.)
-e [float] : 静電相互作用の項の重み倍率 (デフォルト:1.0)
-d [float] : 脱溶媒和項の重み倍率 (デフォルト:1.0)
-a [float] : rPSC形状相補性のレセプターコアの値 (デフォルト:-45.0)
-b [float] : rPSC形状相補性のリガンドコアの値 (デフォルト:1.0)
-f [1/2/3] : スコア関数の設定
(1: rPSC形状相補性のみ, 2: rPSC+静電, 3: rPSC+静電+脱溶媒和, デフォルト:3)
-h : ヘルプを表示
予測した複合体の構造の情報はdocking.outというファイルに書き込まれます(デフォルトではスコアの良いものから2000個分).正確にはこのファイルにはリガンドの相対的な移動情報しか書かれていないので,PDBファイルにするには以下の操作を行います.
$ ./decoygen 1.pdb B.pdb docking.out 1
(./decoygen [候補構造のファイル名] [使用したリガンドPDB] [ドッキング出力ファイル] [出力する候補構造の順位])
これで複合体の(1位の)構造1.pdbができました.
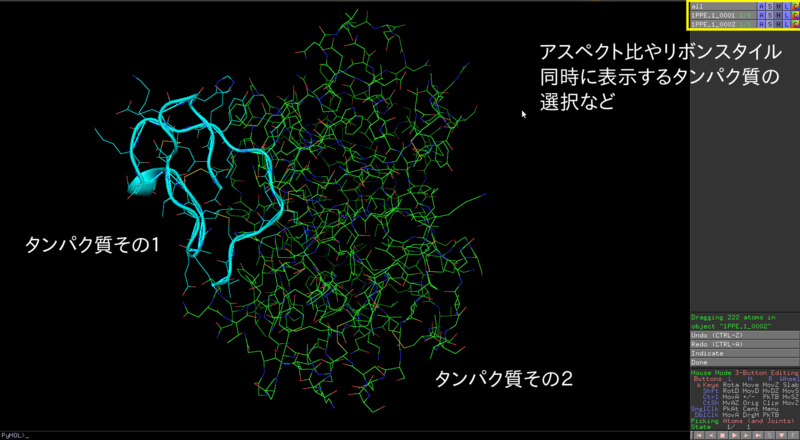
PyMOL (http://pymol.org) などのビューワで実際に見てみましょう.
$ pymol 1.pdb
スレッド並列
MEGADOCKはOpenMPでスレッド並列化されています.
$ export OMP_NUM_THREADS=2
などで適当なスレッド数を指定してあげると速く実行できます.
GPUで動かす
Makefileの編集のときに USE_GPU := 1 とするとGPUで計算するバイナリを生成できます.
かなり高速に計算できますので,是非GPUを使ってやってみてください.
参考情報
- MEGADOCKのHP http://www.bi.cs.titech.ac.jp/megadock/
- 生物物理夏の学校2013 計算ハンズオンセミナー http://www.bi.cs.titech.ac.jp/~ohue/bps2013/