さて、今回はいよいよ動力学計算を行っていきます。
今回扱うテーマはSiの加熱です。前回と同じFeの結晶(α鉄)でもよかったのですが、Si結晶(ダイヤモンド構造)のほうが疎な構造なので可視化したときの見た目がわかりやすいというのがもっぱらの理由です。
動力学計算のスクリプト
はじめにスクリプトを載せておきます。動作させるには前回と同じようにSi.tersoffポテンシャルファイルが必要です。あと結果出力用にcfgディレクトリを作っておいてください。
atom_style atomic
units metal
boundary p p p
lattice diamond 5.4
region region1 block 0 3 0 3 0 3 units lattice
create_box 1 region1
create_atoms 1 box
pair_style tersoff
pair_coeff * * Si.tersoff Si
mass 1 28.0855
thermo_style custom step etotal temp lx vol press
thermo 10
fix f1 all box/relax iso 0.0
minimize 1.0e-10 0.0 1000 100000
unfix f1
reset_timestep 0
timestep 0.001
dump d1 all cfg 10 cfg/run.*.cfg mass type xs ys zs id type
dump_modify d1 element Si
variable energy equal etotal
variable temperature equal temp
fix fout1 all ave/time 1 100 100 v_energy v_temperature file out_energy.txt
velocity all create 2500 12345 dist gaussian
fix f1 all nvt temp 2500.0 2500.0 0.1
run 10000
unfix f1
comm_modify cutoff 8.01
compute distribution all rdf 50 1 1 cutoff 6.0
fix fout2 all ave/time 10 100 1000 c_distribution[*] file out_rdf.txt mode vector
fix f1 all nvt temp 2500.0 2500.0 0.1
run 1000
主に前回以降に追加されたところを紹介します。その前にまず変更点として、原子が変わっているのでlatticeがダイヤモンド構造に、pair_*Si用のものに変わっています。
minimize以降が今回の追加した分です。まず、動力学計算なので時間刻みの設定(timestepコマンド)が必要です。
dumpコマンドは原子の配置の書き出しに使うコマンドです。様々な形式に対応していますが、ここではAtomeye用のcfgファイル形式を指定しています。後で可視化用に使います。
その後のvariableコマンドは初出です。これはMD計算中の様々な値を保持したり、それらの演算をしたりする際に使えるコマンドです。
variableというコマンド名からプログラミング言語における変数の定義のように感じられますが、これはちょっとミスリーディングな名前だと思います。実際にはvariableはどこかで参照されるたびに内部の値が再計算されます。例えばvariableに温度を代入(?)しておくと、参照するたびにその瞬間の温度が返ってきます。変数というよりは(手続き型言語における)関数というほうがイメージが近いと思います。
variableは原子ごとに値を割り振ることもでき、うまく使うとLAMMPSの自由度が上がります。
fix ave/timeコマンドはrunの間にオプションで指定した値を出力し続けるコマンドです。ここでは"out_energy.txt"というファイルにエネルギーと温度を書き出しています。
velocityコマンドは原子に強制的に初速を与えるコマンドです。はじめが0ケルビンだと全く動かないため、初速を適当に与える意味があります。
fix nvtコマンドは系にNVTアンサンブルを適用するコマンドです。つまり温度制御をかけます。後ろの3つの値は「MD開始時の熱浴の温度」「MD終了時の熱浴の温度」「熱浴の順応性」です。
3つ目の値が少し直感的でない値ですが、曖昧に表現すると(人工的な)熱浴に対する慣性のようなパラメータです。おおよそtimestepで設定した値の100倍くらいにしておけば大丈夫です。(圧力制御の場合は1000倍程度。)
類似のコマンドにfix npt(温度と圧力が一定)やfix nve(体積とエネルギーが一定: 温度も圧力も制御しない)など様々なアンサンブルに対応したコマンドがあります。
制御しない場合に対応するはずのNVEアンサンブルでさえfixコマンドが用意されているのは初見では不思議に見えるかもしれません。
これは、運動方程式の数値積分、つまり原子にかかる力から位置や速度を更新する処理(LAMMPSでは速度ベルレ法)がfix nveに入っているからです。実装を読める人はソースの"fix_nve.cpp"を見てみるのが一番早いと思います。
逆にいうと、これらアンサンブルのfixのいずれかを定義しないと原子は全く動きません。fixなしでrunすると、これでは原子が動かないぞと警告が表示されます。
スクリプトの解説に戻ると、run 10000がこのシミュレーションのメインループです。
その後のcomputeコマンドと短いrunコマンドは動径分布関数を"out_rdf.txt"に保存するためのおまけです。とりあえずここでは説明は省略します。(消しても動きます。)
MD計算自体は1コアでもすぐ終わると思います。
実行結果の見方と可視化
さて、実行結果を見てみましょう。まずはエネルギーを出力した"out_energy.txt"からです。
gnuplot
>> plot "out_energy.txt" with lines
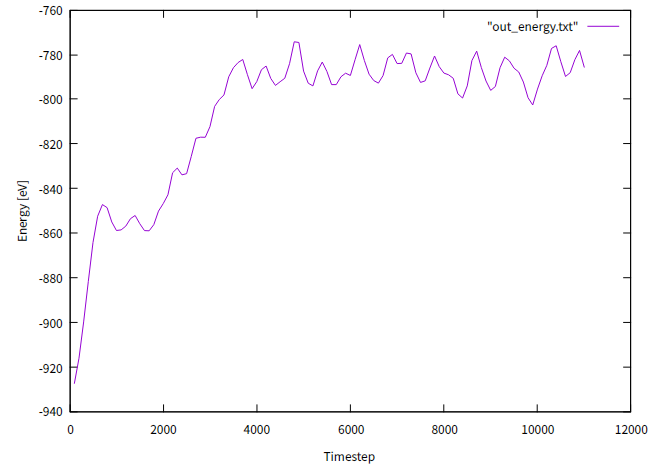
はじめの4000ステップくらいはエネルギーが増加していく遷移状態であることがわかります。4000ステップのあたりからおおよそエネルギーが安定しています。
これはつまり、はじめの4000ステップ中は目的としているNVTアンサンブルではなく初期条件に依存した状態だということです。
このような緩和計算はMD計算を行う際に一般的に必要とされるプロセスです。この範囲で(平衡状態に達していると勘違いして)何かMD計算を行うと間違った結果が得られてしまいます。
遷移状態と書きましたが、この範囲で何がおきているのかは実際に原子の動きを見てみるとよくわかります。Atomeyeを使って可視化してみます。
A cfg/run.0.cfg

Atomeyeの操作は独特です。キーボードのdelete, insert, home, end, pageup, pagedownのあたりのキーで操作します。その他bでボンド、lとkで配位数と原子種の切り替えなど各キーが表示内容の指示と対応しています。
上に0ステップ、2000ステップ、10000ステップのときのそれぞれのAtomeyeの出力を載せています。構造を見てみるとわかりますが、最終的に溶けていることがわかります。(2500ケルビンなのでSiは実際に溶けるでしょう。)
ただし、溶けるまでしばらく時間がかかっています。これが先のエネルギーのグラフにおける緩和計算が必要な時間と対応しています。
おまけで出した動径分布関数もGnuplotで描画できます。
gnuplot
>> plot "out_rdf.txt" using 2:3 with lines
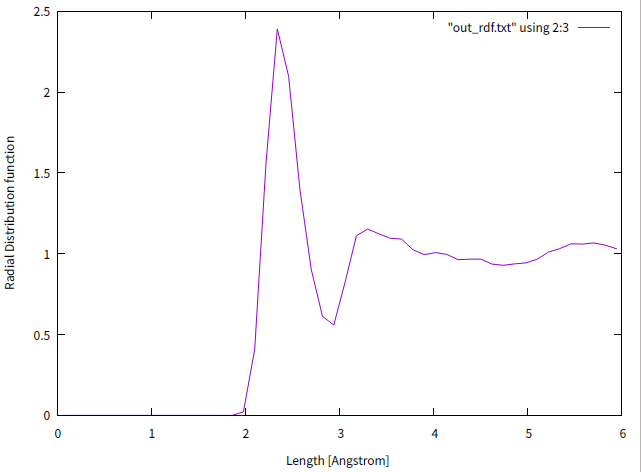
ピークが広がり、第二近接原子以降はなめらかな曲線になっています。ここからも溶けていることがわかります。
次回はシミュレーション結果の保存やコマンドの制御について紹介します。
← LAMMPS入門の手引き 1:静的な計算
→ LAMMPS入門の手引き 3:データの保存と制御文
応用課題
-
この溶けた状態からゆっくり冷却していくとアモルファス構造が得られる。アモルファス構造の動径分布関数を溶けた状態の動径分布関数と比較してみよう。実験結果とも比較してみよう。
-
MD計算のごく初期をよく見ると、開始時に2500ケルビンあった温度が一度1000~1500ケルビンまで下降している。この理由は何だろうか。
-
もしNVTアンサンブルでなくNPTアンサンブルを使うとどうなるだろうか。