前回の記事はPython版のCaImAnの使い方について説明しました。
今回の記事では、MATLAB版のCaImAnの使い方から内部のアルゴリズムの中身、さらに取り出したカルシウム信号をスパイクレートに変換するデコンボリューションのコード実行まで紹介します。
MATLAB版のCaImAnのgithubページ(wikiあり)(https://github.com/flatironinstitute/CaImAn-MATLAB (wiki))
論文(https://elifesciences.org/articles/38173)
MATLAB版のCaImAnのインストールは簡単で、githubからダウンロードしてそこにパスを通すだけです。
「パスの設定」などでCaImAn-MATLAB-masterをパスに追加しておきましょう。
または、MATLABのコマンドウィンドで
addpath(genpath('<CaImAn-MATLAB-masterのパス>'));
でもパスの追加ができます。
またCaImAnの前提として、以下のツールボックスをインストールする必要があります。(③と④は無くても動くが機能を全て使うには必要)
①Statistics and Machine Learning Toolbox
②Image Processing toolbox
③Parallel Computing toolbox
④Deep Learning Toolbox(昔のNeural networks toolbox)
ちなみにstudent suiteでは、④は含まれていませんので、購入する必要があります。
また、④の機能をつかうためには、無料のDeep Learning Toolbox Importer for TensorFlow-Keras Modelsも必要のようです。(https://jp.mathworks.com/matlabcentral/fileexchange/64649-deep-learning-toolbox-importer-for-tensorflow-keras-models)
CaImAnの内部アルゴリズム
CaImAnの内部アルゴリズムは以下の2段階に分けられます。
1.CNMF(条件付き非負値行列因子分解)によって点滅領域(細胞候補領域)の取り出し
2.1で得られた細胞候補領域から細胞候補を選別する
なぜ二段階に分かれているのかというと、1のCNMFは指定した数の領域を必ず取り出すアルゴリズムだからです。つまり入力動画にどれくらい細胞があるかにかかわらず指定した数だけ領域を取り出します。
細胞数の少ない入力動画では無理やり背景領域からの点滅を取り出されてしまうことが発生します。
またCNMFでは、点滅のパターンが同じピクセルを取り出すだけで細胞の形は考慮していません。(細胞の大きさは考慮される)
そのため、2でCNN(畳み込みニューラルネットワーク)を用いることで、その領域が細胞であるかを判定する仕組みになっています。
以下に処理の詳細を示します。
1.CNMF(条件付き非負値行列因子分解)によって点滅領域(細胞候補領域)の取り出し
画像から情報を取り出す手法としてNMFが有名ですが、これは行列分解をすることで教師なしで特徴量を抽出することができます。
これを細胞領域を取り出す場合に応用していて、NMFを用いて同じような輝度変化をしているピクセルで取り出していけば、細胞領域を取り出すことができるというわけです。
しかし、今回は細胞領域を取り出す性質上、取り出される領域は多くのピクセルに渡らず局所に固まっていなければなりません。これを実装するために、空間情報にL1正則化を加えて条件付きのNMF(CNMF)を行っているようです。
(詳しいアルゴリズムは論文https://doi.org/10.1016/j.neuron.2015.11.037 を参照)
数式のイメージとしては以下のようになります。
Y = A*C + B + E
Yは、動画のxy方向を一方向に並べることで、動画を2次元行列として表現したものです。(ピクセル数×フレーム数)
Aは、細胞の空間情報になります。(ピクセル数×細胞数)
Cは、細胞の輝度変化の値になります。(細胞数×フレーム数)
Bはバックグラウンドの輝度です。Eは誤差でこれを最小にするようにします。
ただしこのNMFやCNMFでは、細胞数(特徴量の基底の数)はパラメータとして人が決める必要があります。
細胞の数を自分で正確に求めることは困難であるため、CaImAnではこの細胞の数を多めに設定してとりあえず抽出しておいて、次の段階で抽出された領域が本当に細胞であるかを判断しているようです。
2.得られた細胞候補領域から細胞を選別
この段階では、1で抽出された領域が細胞であるかを判定しています。
判定は3つの基準で行われています。
1.その領域内のピクセルのコリレーションを計算し、一定よりも高いかどうか
2.その領域の輝度変化のSN比が一定よりも高いか(つまり一回でも細胞の活動(一過性の輝度上昇)がみられるかどうか)
3.CNNを用いてその細胞の形態を評価し、その値が一定より高いかどうか
この3点の閾値はパラメータとなっており、変更することで結果も変わります。
ちなみに3つ目をするにはDeep Learning toolboxが必要になります。
CaImAnの使い方とパラメータ調整
フォルダ内のdemoscript.mが実行スクリプトになります。
これを実行することで、デモの動画から細胞領域を取り出すプログラムを動かすことができます。
まずはこれがエラー無く動くかどうかを確認しましょう。
自分の動画を入力にしたい場合は7行目の変数nanにそのパスを入れましょう。
nam = '<tiffスタック形式の動画のパス>';
以下にdemoscript.mの中で調整するべきパラメータと変更するべき場所を紹介します。
パラメータ
20行目のK・・・これはCNMFの細胞(の候補領域)の数に当たります。目算での細胞数の少し多めの値を入れましょう。
21行目のtau・・・これはCNMFの正則化の度合い決める値になります。細胞のピクセルスケールでの半径にしましょう。
30行目のmin_SNR・・・これは細胞選別に用いる2つ目の基準の閾値です。
31行目のspace_thresh・・・これは細胞選別に用いる1つ目の基準の閾値です。
32行目のcnn_thr・・・これは細胞選別に用いる3つ目の基準の閾値です。
コード修正箇所
71~75行目のtry-catch構文は、Deep Learning toolboxがない場合でもエラーにならないようにするためのものです。toolboxがある場合は修正して必ずcnn_classifierを使うようにする方がよいです。
84行目では、このままだと3つの基準のうち1と3はどちらかを満たせばよいという記述になっています。
3つ基準全てをクリアしたものだけを細胞として選びたい場合は、
keep = (ind_corr & ind_cnn) & ind_exc;
このように修正しましょう。
このほかにもパラメータなどはありますが、基本的にはデフォルトの値で良いかと思われます。
demoscript.mの後は、postProcessCNMF.mで結果の可視化もできます。これをするには
以下のコマンドを実行して変数のタイプを整えおけばできます。
A2 = full(A2);
Yr = double(Yr);
postProcessCNMF
結果として以下の図が表示されました。

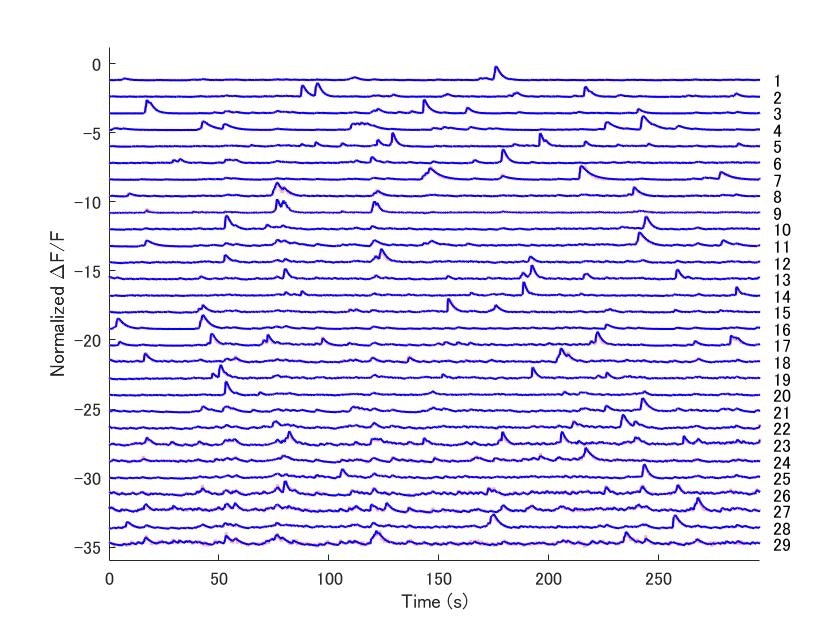
デコンボリューション
CaImAnの関数の中にはデコンボリューション機能があります。デコンボリューションとは、細胞の輝度変化から細胞の活動を推定する方法になります。カルシウムイメージングでは、細胞にカルシウム感受性蛍光タンパク質発現させて行われますが、その時定数などの影響を受けて輝度変化は生成されます。また輝度変化の中にはノイズも多少含まれています。デコンボリューションによってそれらの影響を取り除くことができるとされています。
関数の詳細はこのサイトにあります。(https://github.com/zhoupc/OASIS_matlab)
demoscript.mは細胞領域の取り出しまでで、デコンボリューションまではしてくれません。run_pipeline.mで一連の機能を使えるはずですがNoRMCorreSetParms関数がないためエラーになりました。
そのため、自分で関数を使うことにしました。
デコンボリューションの関数の中にはCVX libraryを必要とするものもあるのでインストールしておきます。postProcessCNMF.mした後の変数raw.dfofにはdF/F値が含まれていると思われるのでそれをデコンボリューションしてみます。
n = 1;
[c, s, options] = deconvolveCa(raw.dfof(n,:));
nは細胞の番号です。
出力されたcはノイズが消された輝度変化、sがデコンボリューション得られたスパイクレートです。
デコンボリューションにはオプションがあり、それを変更することで結果も変化します。
図にして表示してみましょう。
figure
subplot(2,1,2)
plot(raw.dfof(n,:))
hold on
plot(c)
title('calcium trace')
legend('raw trace','denoised trace')
subplot(2,1,2)
plot(s)
title('spike rate')
以下のような図になりました。
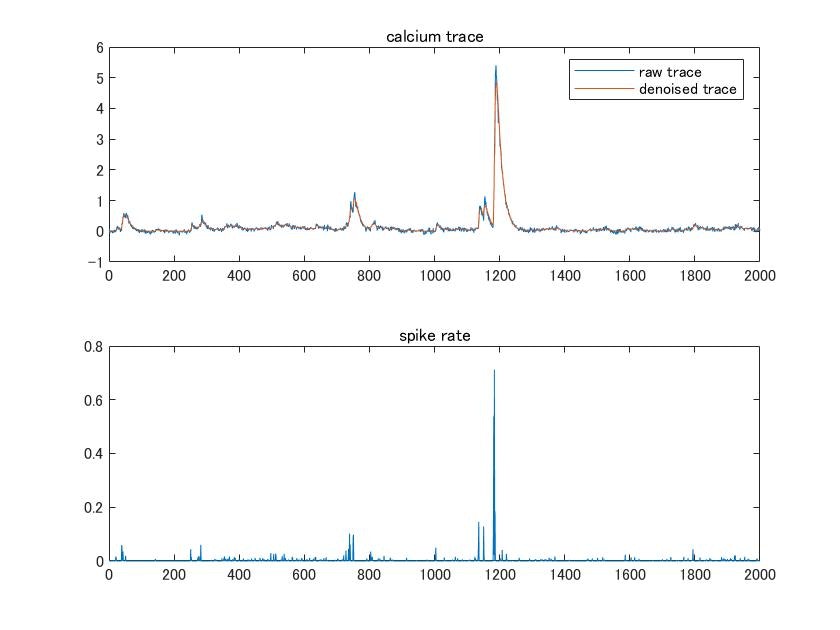
こんな感じでデコンボリューションによって、denoised traceと、spike rateが得られます。
終わりに
CaImAnの機能はまだまだあるようで、例えばサイズの大きな入力動画に対してはタイル分割して処理する機能があったり、GUIで解析できる機能があったり、3次元カルシウムイメージング(つまりxyztの4次元の動画)に対応する機能のあるようです。ほかにも複数日の動画をつなぎ合わせて同一細胞を追う機能や、モーションコレクションをしてから行うパイプライン機能もあるようです。
追記:一連の機能を使う方法がわかりました。別記事にしてあります。
https://qiita.com/takeajioka/items/2d25ea7b64c59de3c2e6