Mygeneを使った遺伝子情報の取得
遺伝子名やデータベースのアクセッションナンバーなどから遺伝子情報を引き出す時のパッケージとして、以前はRのBiomaRtを利用していましたが、リンク切れが多発していることや、データベースへの問い合わせに時間がかかったり、わざわざsqlliteドライバーを作るなど一手間かけないといけないことや、関数が多すぎて使いづらいという気持ちでいました。そこで久々に色々なannotation packageを調べていたのですが、mygeneというpythonとRで実装されているサービスを発見したので、pythonでの使い方を簡単に紹介したいと思います。
mygeneが何者かというのは、ウェブサイトを拝見していただければと思うのですが、約30種類ほどのIDをクイック変換できるサービスで、他のannotation toolに比べて更新も頻繁で、シンプルで使いやすいと思います。
環境
- OSX High Sierra 10.13.3
- Python 3.6.0 |Anaconda 4.3.1 (x86_64)| (default, Dec 23 2016, 13:19:00)
- [GCC 4.2.1 Compatible Apple LLVM 6.0 (clang-600.0.57)]
mygeneのインストール
pip install -e git+https://github.com/sulab/mygene.py#egg=mygene
テストクエリー用の遺伝子シンボルの取得
今回はQueryとして、遺伝子シンボルを使いたいと思います。本来出発点としては、名前が変更されうる遺伝子シンボルよりもEntrezIDなどの方が良いですが、NAが現れるという意味ではいい例題です。もちろん、他のIDも利用できます。適当な遺伝子シンボルセットを取得するためにGSEAのDBであるMsigDBからsymbol listを得ました。メールアドレスを登録すれば、取得することができます。
取得したテキストファイルをパースします。 mygeneに入力するために改行された文字列をlist構造にしています。csv.readerを使い、改行を区切り文字として読み込み、for文を使って中身を1つのリストに結合させています。その際に2行のheader情報がじゃまなので、itertoolsのislice関数を使って2行目まで読み飛ばしています。
import csv
from itertools import islice
msig_file = "geneset.txt"
sl = list()
with open(msig_file, 'r') as fl:
reader = csv.reader(fl,delimiter='\n')
for row in islice(reader, 2, None):
sl.extend(row)
print(sl)
slオブジェクトの中身
['ADAM10', 'ADAM17', 'AGXT', 'ANGPT4', 'ANXA4', 'APH1A', 'APH1B', 'APP', 'ASCL1', 'ATOH1', 'BMP2', 'C12orf52', 'CDH6', 'CDK6', 'CDKN1B', 'CEBPA', 'CHAC1', 'CNTN1', 'CNTN6', 'CREBBP', 'DLL1', 'DLL3', 'DLL4', 'DNER', 'DTX1', 'DTX2', 'DTX3', 'DTX4', 'EP300', 'EPN1', 'ETV2', 'FCER2', 'FOXA1', 'FOXC1', 'FOXC2', 'GALNT11', 'GMDS', 'GOT1', 'HES1', 'HES5', 'HES7', 'HEY1', 'HEY2', 'HEYL', 'HHEX', 'HNF1B', 'HOXD3', 'IFT172', 'IFT74', 'IL2RA', 'ITCH', 'ITGB1BP1', 'JAG1', 'JAG2', 'KAT2B', 'KCNA5', 'KRT19', 'MAML1', 'MAML2', 'MAML3', 'MDK', 'MESP1', 'MESP2', 'MIB1', 'MIB2', 'MYC', 'NCSTN', 'NEURL', 'NEURL1B', 'NEUROD4', 'NKAP', 'NLE1', 'NOTCH1', 'NOTCH2', 'NOTCH2NL', 'NOTCH3', 'NOTCH4', 'NR0B2', 'NR1H4', 'NRARP', 'ONECUT1', 'PERP', 'PGAM2', 'PLN', 'POFUT1', 'PSEN1', 'PSEN2', 'PSENEN', 'PTP4A3', 'RBM15', 'RBPJ', 'RIPPLY1', 'RIPPLY2', 'RPS19', 'RPS27A', 'S1PR3', 'SEL1L', 'SNAI1', 'SNAI2', 'SNW1', 'SOX9', 'SPEN', 'SUSD5', 'TBX2', 'TGFB1', 'TGFBR2', 'TIMP4', 'TMEM100', 'TP63', 'UBA52', 'UBB', 'UBC', 'WDR12', 'ZNF423']
クエリーの準備ができましたので、mygeneを使ってみたいと思います。mgというインスタンスを作り、queryを沢山読み込むのでquerymanyメソッドを使います。speciesを指定しなければ、1つの遺伝子につき複数のオーソログの情報が出力され、too much informationになるので指定しておきます。
import mygene #mygene
mg = mygene.MyGeneInfo()
res = mg.querymany(sl, 'name,symbol',as_dataframe=True,species='human')
結果のテーブル
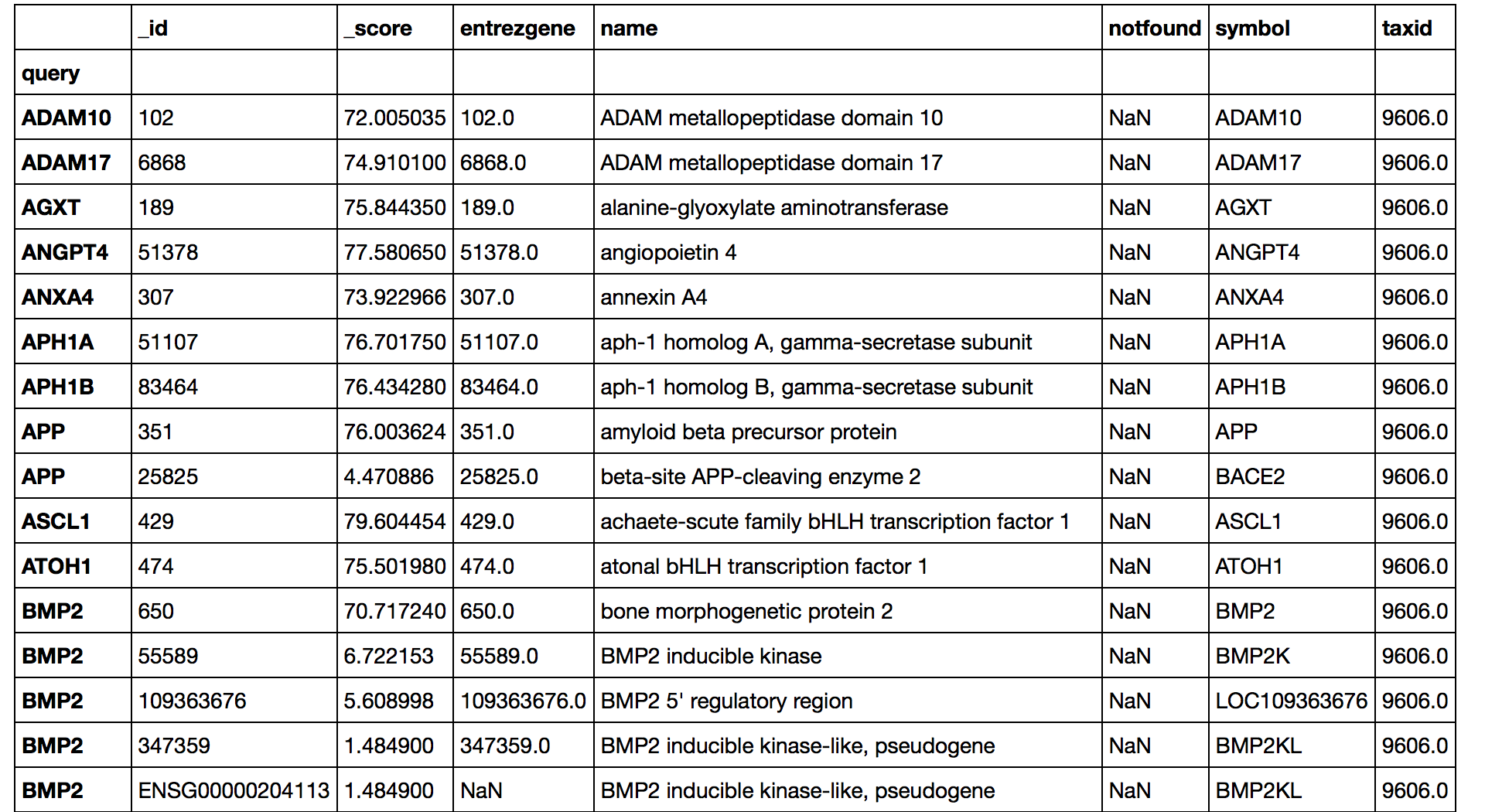
この様に遺伝子情報のテーブルを簡単に作ることができます。しかも速いです!
他の使い方については、本家のウェブサイトよりもPythonのドキュメントページが分かりやすいと思いますので、是非ご覧になってみてください。