本稿では、Pythonを用いRNA-Seq (シングルセルではなくバルク) データから、サンプル間の遺伝子発現様態の特徴を可視化するための、UMAPを実行し、グラフに出力する方法を解説する。
UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction
参考: https://umap-learn.readthedocs.io/en/latest/ (2022年6月9日アクセス)
UMAPとは次元削減の手法のひとつである。他の次元削減としてPCA (Principle Component Analysis) や t-SNE (t-Distributed Stochastic Neighbor Embedding) が有名であるが、PCAはRNA-Seqデータのような比較的大きいデータに適用するとうまく特徴を示してくれないことがある。また、t-SNEはUMAPとともに高次元データの次元削減手法として有名であるが、UMAPより低速であることが知られている。本稿ではUMAPの原理は詳しく解説しないので、原著論文や解説をしているウェブサイトを参照してほしい (原著論文, McInnes et al. arXiv 2018)。
Pythonを用いたUMAP
使用データ
本項では、先行研究の公開データを用いてデモ解析を行う。
生後1ヶ月、2ヶ月、4ヶ月齢の雌雄両方のマウス由来の海馬を用いたRNA-Seq解析データを利用する (Bundy et al. BMC Genet. 2017, #GSE83931)。
| Age | Sex | Sample Number |
|---|---|---|
| 1 month | Male | 5 |
| 1 month | Female | 5 |
| 2 month | Male | 5 |
| 2 month | Female | 5 |
| 4 month | Male | 5 |
| 4 month | Female | 5 |
上記公開データのXlsxファイル(GSE83931_Counts_and_FPKM.xlsx)の中の「FPKM」シートのデータを解析に使用しているが、"Associated.Gene.Name"と"Description"のカラムは削除してからUMAP解析に使用した。
データ例:
| GeneID | 1071D | 1071E | ・・・ | 1056G | 1056H |
|---|---|---|---|---|---|
| Gene_A | 1.2 | 1.1 | ・・・ | 2.2 | 3.2 |
| Gene_B | 1.0 | 0.9 | ・・・ | 1.1 | 1.1 |
| ・・・ | ・・・ | ・・・ | ・・・ | ・・・ | ・・・ |
| Gene_Y | 321.4 | 123.4 | ・・・ | 234.5 | 345.6 |
| Gene_Z | 0.0 | 0.0 | ・・・ | 0.0 | 0.0 |
Pythonでの実装
UMAPにおいてはパラメータを変更することによりデータの見え方が大きく変わるが、本項では特に重要な3つのパラメータ、n_neighbors, min_dist, metricを変更しながらデータの見え方の最適化を行うスクリプトを紹介する。
#使用モジュールのインポート
import numpy as np
import pandas as pd
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import StandardScaler
import umap
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
import seaborn as sns
%matplotlib inline
filepath = 'ここにファイルパスを記入'
rsdata = pd.read_excel(filepath, sheet_name='FPKM' ,header=0, index_col=0)
rsdata.dropna(how='any', inplace=True) #公開データ内に欠損値があったので削除
rsdata = rsdata.T #データの転置
rsdata_val = rsdata.values #データをnumpy arrayに変換
scaled_rsval = StandardScaler().fit_transform(rsdata_val) #データの標準化
上記までがデータの下準備で、下記スクリプト内のdraw_umap関数が実際にumapを作成する機能をもつ部分である。
#グラフの体裁を整えるための準備
red1 = [1.0, 0.57, 0.57]; red2 = [1.0, 0.86, 0.86]
blue1 = [0.57, 0.57, 1.0]; blue2 = [0.86, 0.86, 1.0]
#下記 colors配列ではグラフのドットの色を指定しており、の要素数がサンプル数と一致している。
#配列の 先頭->後尾 に向かって、サンプルファイルの 左->右 の色が指定される。
colors = [blue2, blue2, blue2, blue2, blue2, \
red2, red2, red2, red2, red2, \
blue1,blue1,blue1, blue1, blue1, \
red1, red1, red1, red1, red1, \
'blue', 'blue', 'blue', 'blue', 'blue', \
'red', 'red', 'red', 'red', 'red']
def draw_umap(n_neighbors=15, min_dist=0.1, n_components=2, metric='euclidean', title=''):
fit = umap.UMAP(
n_neighbors=n_neighbors,
min_dist=min_dist,
n_components=n_components,
metric=metric
)
u = fit.fit_transform(scaled_rsval);
plt.rcParams['font.family'] = 'Helvetica' #グラフ内のフォントの変更
plt.rcParams['lines.linewidth'] = 0.5
fig = plt.figure(dpi=200, figsize=(3,3))
if n_components == 1:
ax = fig.add_subplot(111)
for i in range(len(u)):
ax.scatter(u[:, 0][i], c=colors[i], marker=markers[i])
if n_components == 2:
ax = fig.add_subplot(111)
for i in range(len(u)):
ax.scatter(u[:, 0][i], u[:, 1][i], color=colors[i], s=60, edgecolors='black')
if n_components == 3:
ax = fig.add_subplot(111, projection='3d')
for i in range(len(u)):
ax.scatter(u[:, 0][i], u[:, 1][i], u[:, 2][i], c=colors[i], marker=markers[i], s=20)
plt.title(title, fontsize=18)
n_neighbors
まず初めに、n_neighborsのパラメータを試す。n_neighborsはデータのlocalの特徴か、globalの特徴をみるかのバランスを決める。n_neighborsが小さいときはよりlocalに、大きいときはよりglobalに着目する。
for n in (2, 3, 4):
draw_umap(n_neighbors=n, title='n_neighbors = {}'.format(n))
plt.savefig('Figures/UMAP_neighbors'+str(n)+'.png')

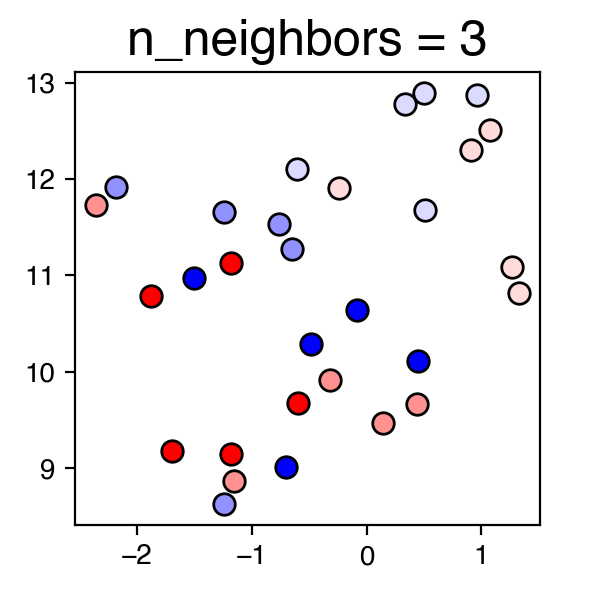
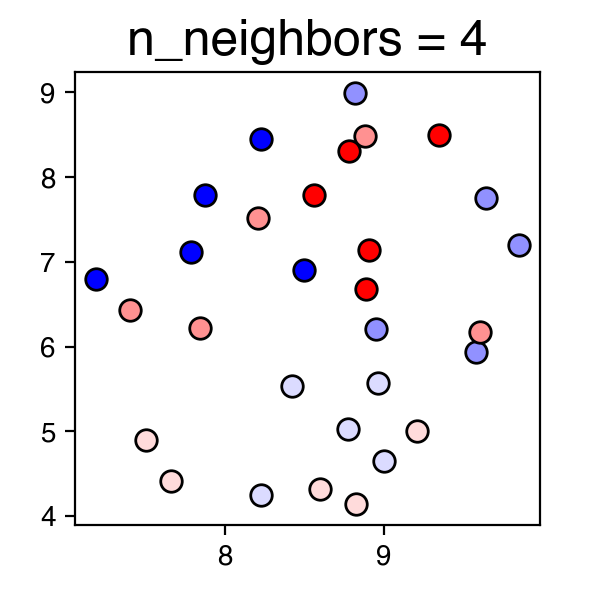
上記の結果グラフにおいて、青・赤の点はそれぞれオス・メスのサンプルを示しており、それぞれの色が濃くなるほど月齢が大きいサンプルとなっている。
今回はn_neighbors=3が良い感じがするので、n_neighbors=3に固定して、次のパラメータに進む。
min_dist
min_distは点同士をどれくらい近くにクラスターするかのバランスを制御する。すなわち、min_distが小さい場合、クラスタリングの結果はよりlocal・タイトになる。対してmin_distが大きい場合はより全体の特徴を保持するようクラスタリングされる。
for d in (0.0, 0.1, 0.2, 0.4, 0.6, 0.8):
draw_umap(n_neighbors=3, min_dist=d, metric='euclidean', title='min_dist = {}'.format(d))
plt.savefig('UMAP_neighbours3_mindist_'+str(d).replace('.', '')+'_euclidean.png')
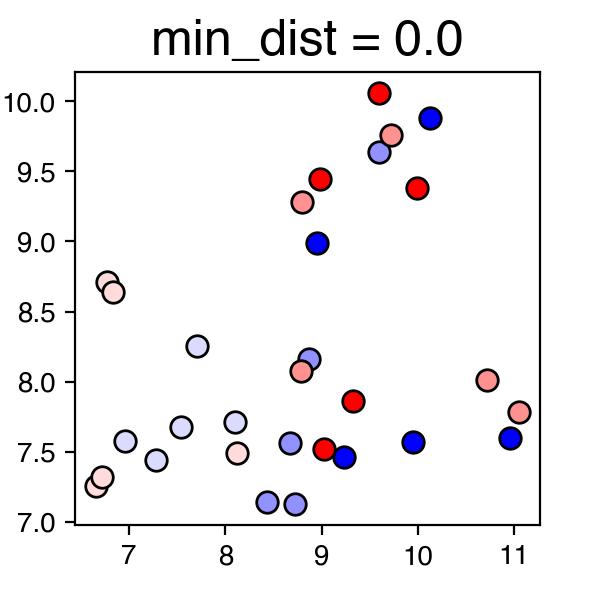



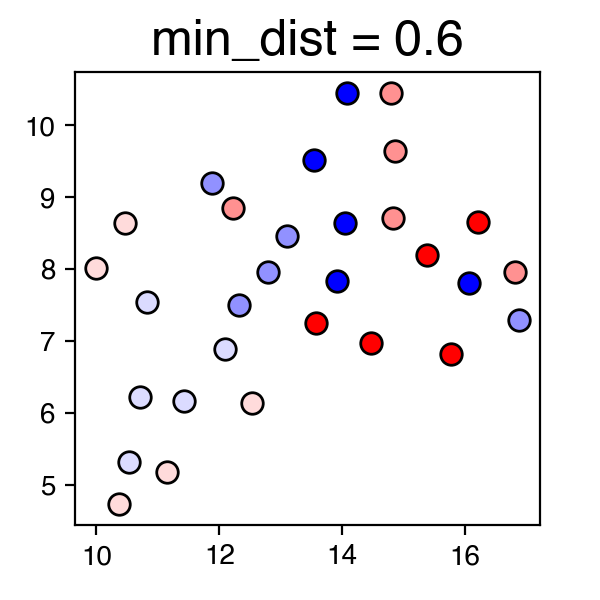

データによってはmin_distは割とデータの見た目に効いてくるので、自分のデータから主張したいことをサポートするようなものを得られるよううまく調整する。
この結果の中だとmin_dist=0.1が一番良い感じがするので、min_dist=0.1に固定して本稿最後のパラメータ調整に移る。
metric
metricは距離の計算方法を指定するパラメータである。ここの算出方法については詳しく解説しないが、これも結構見た目に効いてくる。
for m in ['euclidean', 'manhattan', 'chebyshev', 'minkowski', 'canberra', 'braycurtis']:
draw_umap(n_neighbors=3, min_dist=0.1, metric=m, title='metric = {}'.format(m))
plt.savefig('UMAP_neighbours3_mindist01_'+str(m)+'.png')
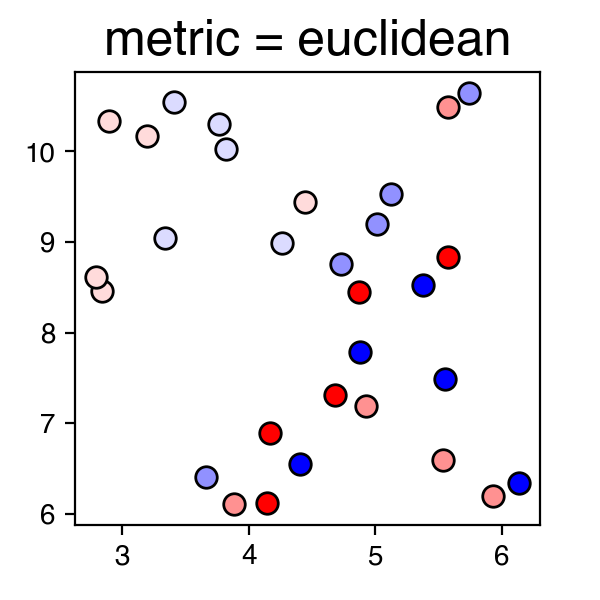
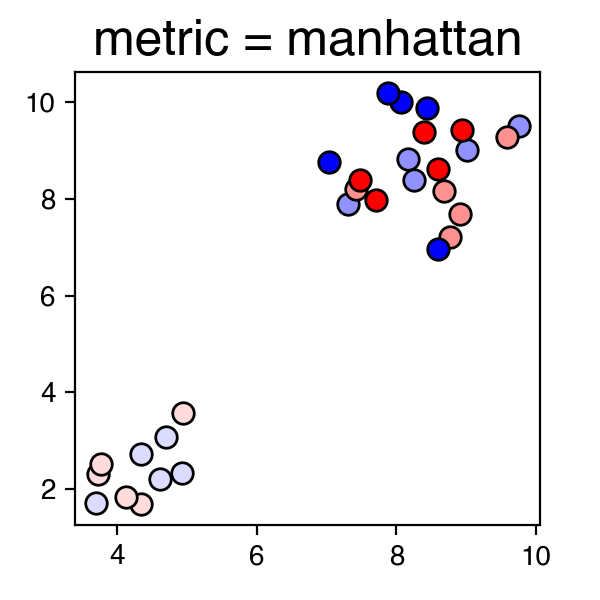

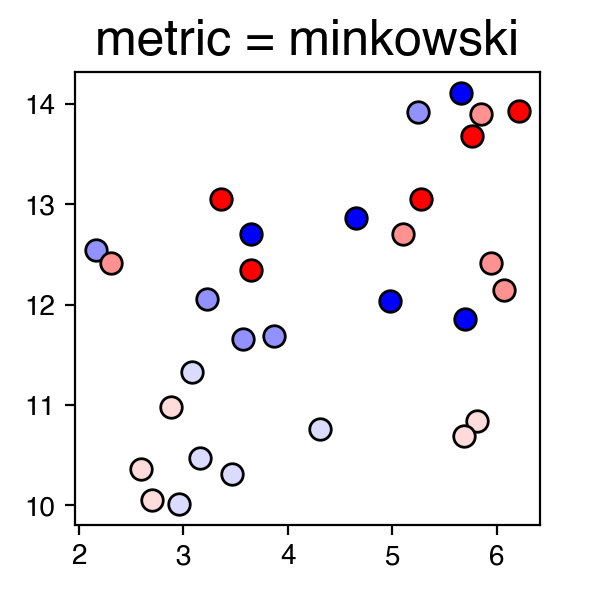
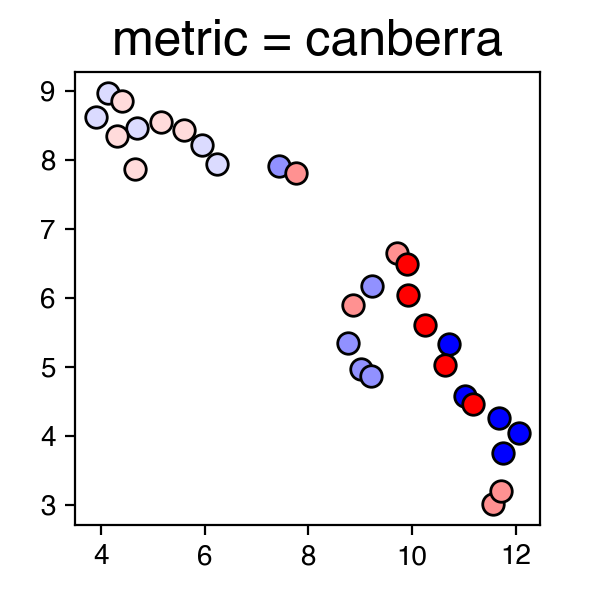
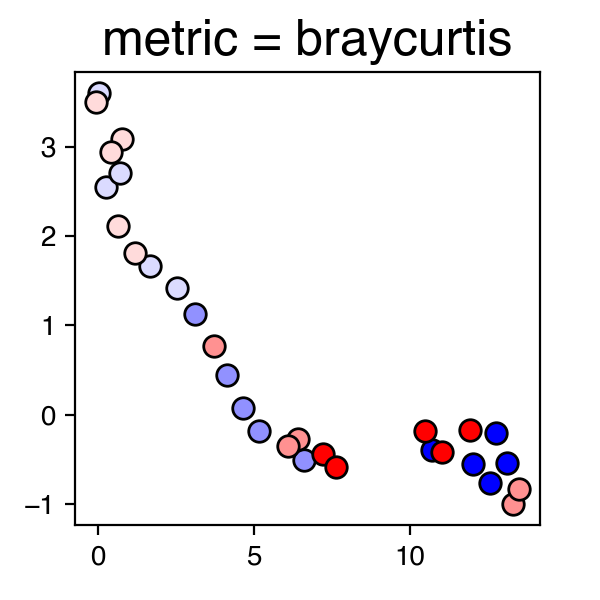
上記の結果を見ると、"manhattan", "canberra", "braycurtis"あたりが良さそうである。今回用いた公開データの詳細については原著論文を確認していただきたいが、今回得られた結果「のみ」から推察すると、特に"canberra"や"braycurtis"において、グラフ左から右にかけて、サンプルのステージ (月齢) が上がっていく様子がみられ、腑に落ちる結果であると感じる。またオス・メスに着目すると、1ヶ月齢のサンプルではオス・メスが雑然と混じっているが、2ヶ月・4ヶ月齢のサンプルにおいてはオス・メスが比較的分かれてクラスタリングされているようにみえる。
おわりに
本稿では公開データを用いて、RNA-Seqデータに対してUMAPのクラスタリングを行う方法を解説した。今回、PythonのUMAP公式ディスクリプションのサイト(https://umap-learn.readthedocs.io/en/latest/parameters.html, 2022年6月9日アクセス)を参考として、n_neighbors, min_dist, metricの順でパラメータを変更・固定して行ったように説明したが、実際にはn_neighbors, metric, min_distの順番でパラメータを決めていった。今回の結果を見ていただいてわかるように、UMAPを実行するスクリプトはシンプルで実行時間もそれほどかからないが、パラメーターの組み合わせによって見た目が大幅に変わってくるので、自分の仮説になるべく沿うようなグラフを得られるよう、自分で様々な組み合わせを試してほしい。
(ちなみに、n_components=3を指定するとUMAPの結果が3次元で出力されるのでお時間のある方は試してみてほしい)。