本稿では、Pythonを用いRNA-Seq (シングルセルではなくバルク) データから、サンプルの遺伝子発現様態の特徴を可視化するための階層的クラスタリングの実行、およびヒートマップにより可視化する方法を解説する。
階層的クラスタリング
参考: https://bcblog.sios.jp/what-is-clustering-merit-demerit/ (2022年6月14日アクセス)
クラスタリングとは、ある集合を何らかの規則によって分類することである。言い換えると、RNA-Seqデータの場合では「サンプル間の (遺伝子間の) 遺伝子発現パターンが似ているか否か」という類似性に基づいてグループ分けする作業である。
使用データ
本稿では、先行研究の公開データを用いてデモ解析を行う。
生後1ヶ月、2ヶ月、4ヶ月齢の雌雄両方のマウス由来の海馬を用いたRNA-Seq解析データを利用する (Bundy et al. BMC Genet. 2017, #GSE83931)。こちらの公開データのXlsxファイル(GSE83931_Counts_and_FPKM.xlsx)の中の「FPKM」シートのデータを解析に使用しているが、"Associated.Gene.Name"と"Description"のカラムは削除してからUMAP解析に使用した。
データ例:
| GeneID | 1071D | 1071E | ・・・ | 1056G | 1056H |
|---|---|---|---|---|---|
| Gene_A | 1.2 | 1.1 | ・・・ | 2.2 | 3.2 |
| Gene_B | 1.0 | 0.9 | ・・・ | 1.1 | 1.1 |
| ・・・ | ・・・ | ・・・ | ・・・ | ・・・ | ・・・ |
| Gene_Y | 321.4 | 123.4 | ・・・ | 234.5 | 345.6 |
| Gene_Z | 0.0 | 0.0 | ・・・ | 0.0 | 0.0 |
Pythonでの実装
seabornライブラリのclustermapを用いる。file_pathはデータのパス
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import math
data = pd.read_excel(file_path, sheet_name='FPKM', header=0, index_col=0)
以下ではクラスタリングを適用するために、データを整える作業を行なっている。欠損値があるとクラスタリングを行なってくれず、また発現量が低すぎる遺伝子が残っていると、そっちに引っ張られてヒートマップが一色になってしまう。
#欠損値があったので、削除
data.dropna(how='any', inplace=True)
# 発現量が低すぎる遺伝子を削除。今回はFPKM < 0.001の遺伝子を削除しているが、任意の値を設定すること
targets = []
for i in data.index:
for c in data.columns:
if data.at[i, c] < 0.001: targets.append(i)
targets_set = set(targets)
data.drop(targets_set, inplace=True)
以下ではグラフ作成の細かい準備を行なっている。ヒートマップで使用する色はデフォルトで存在しているカラーマップ (https://pod.hatenablog.com/entry/2018/09/20/212527 2022年6月17日アクセス) も使用可能だが、論文等で使用する場合は自分で色を設定したい場合がある。
# Colormapの作成
from matplotlib.colors import LinearSegmentedColormap
def generate_cmap(colors):
values = range(len(colors))
vmax = np.ceil(np.max(values))
color_list = []
for v, c in zip(values, colors):
color_list.append( ( v/ vmax, c) )
return LinearSegmentedColormap.from_list('custom_cmap', color_list)
cm = generate_cmap(['green', 'orange']) #'green', 'orange'を変更することで、カスタムしたカラーマップを作成できる。
plt.rcParams['font.family'] = 'Arial' #グラフのフォントの設定
以下では、Log2(発現量) を計算する関数および、クラスタリング実行後のデータを返す関数を定義している。
#Log2発現量の計算
def log2calc(d1_data_orig):
d1_data = d1_data_orig.copy()
for i in d1_data.index:
for col in d1_data.columns:
if d1_data.at[i, col] != 0:
d1_data.at[i, col] = math.log2(d1_data.at[i, col])
else:
d1_data.at[i, col] = math.log2(0.001)
return d1_data
#クラスタリング実行後のデータを返す関数
def clust_ID(d2_result, d2_data, filename):
new_index = d2_result.dendrogram_row.reordered_ind
d2_data_c = d2_data.copy()
d2_data_c['tag'] = 0
for i in new_index:
d2_data_c.iat[new_index[i], len(d2_data_c.columns)-1] = i
d2_data_c.sort_values('tag', inplace=True)
d2_data_c.to_excel(filename+'.xlsx')
以下メインスクリプト
data_log2 = log2calc(data)
#col_cluster=Trueにすると、列に対してもクラスタリングを行う
#yticklabelsは行のラベルをつけるか否か
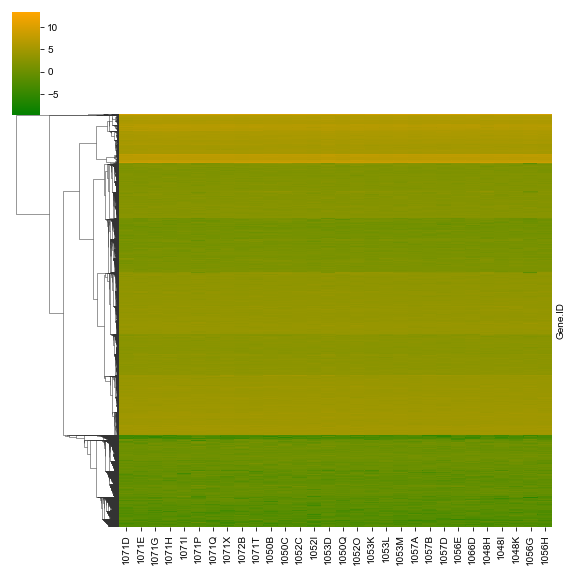
result = sns.clustermap(data_log2, cmap=cm, figsize=(8, 8), col_cluster=False, yticklabels=False)
plt.savefig('Hierarchical_Clustering_Heatmap_Log2FPKM.png')
clust_ID(result, data_log2, 'HClustering')
sns.clustermap()において、z_score=0を指定すると、行に対して Z scoreを算出する。詳しくはウェブ記事を参照してほしいが (http://array.cell-innovator.com/?p=1914 2022年6月17日アクセス)、Z scoreはデータの標準化手法のひとつであり、今回のデータの行に適応すると、「同一サンプル内における、異なる遺伝子間の発現量の差」の情報は無くなるが、「同一遺伝子における、異なるサンプル間の発現量の差」の情報が強調される。
sns.clustermap
result = sns.clustermap(data_log2, cmap=cm, figsize=(8, 8), col_cluster=False, yticklabels=False, z_score=0)
plt.savefig('Hierarchical_Clustering_Heatmap_Log2FPKM_ZScore.png')
clust_ID(result, data_log2, 'HClustering_Zscore')
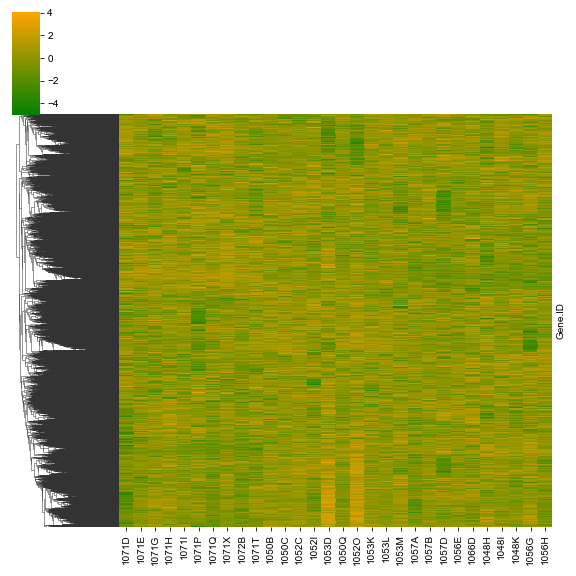
今回は全ての遺伝子に対して実行したが、差次的発現の基準を満たす遺伝子のみに絞りこむ(ANOVAのP値などで)と論文でよく見るような、綺麗なヒートマップになるので試してほしい。
おわりに
本稿では遺伝子間のクラスタリングは行なったが、遺伝子間のクラスタリングは行わなかった。sns.clustermapにおいて、col_cluster=Trueを指定するのみで列に関してもクラスタリングが行えるので、是非試してほしい。ヒートマップの色は各々が他のFigureとぶつからないように調整してしてほしいが、よく見るのは「青・赤」、「緑・赤」、「紫・黄色」などがある。