はじめに
PyTorchベースでグラフ深層学習をするためのライブラリであるDeepGraphLibraryのv0.3.1で, 創薬への応用を意識した機能が追加されたので, 簡単な使用例として分子グラフ生成の方法を紹介します.
今回追加された機能はリリースノートで確認できます.
その中で私が個人的に注目しているのはModel Zoo for Chemistryで, 分子の性質予測と生成・最適化の各種モデル, 訓練コードと学習済みモデルをサポートしています. DGLはGNNs (graph neural networks) の化学への応用に注力しているようですね.
分子生成モデルとしては, いまのところDGMG (deep generative model for graphs)と
JT-VAE (junction tree variational autoencoder) が実装されています.
リリースノートにはDGMGを使ったごく簡単な例が出ているので, ここではJT-VAEで潜在空間での補間 (2つの分子の間を連続的に変化させる) を試してみたいと思います.
準備
主に3つのライブラリが必要になりますのでご用意ください.
- PyTorch: 深層学習フレームワーク
- DGL: PyTorchベースでグラフを扱う深層学習をサポートするライブラリ
- RDKit: 文字列表現から分子グラフを構築したり構造式を描画したりするのに使います
PyTorchとDGLは公式に従えば簡単にインストールできると思いますが, RDKitはやや苦労するかもしれません. 私も詳しくはありませんが, conda環境であれば
$ conda install -c rdkit rdkit
でインストールできると思います.
分子の生成とJunction Tree VAE
即席のイントロです. この分野の知識をある程度お持ちの方は読み飛ばしていただいて構いません.
なぜ分子を生成するのか
薬剤候補となる化合物の数は$10^{23}-10^{60}$との試算がありますが, これらを全て合成して実験するのは現実的ではありません. 毎年新しい化合物が発見されているとはいえ, 現在までに実際に合成されたのは$10^8$程度です [1].
新規化合物を設計したり, その合成方法を考えたり, 実際に合成した化合物を使って実験をしたりと, 創薬のプロセスには莫大な時間とお金がかかるので, AIで効率化できたら嬉しいというのがAI創薬の元々のモチベーションとしてあります.
本記事で扱う分子の生成は, 創薬のプロセスの中でも「薬剤候補となる化合物の提案」に当たるものです.
「この病気に効いて」「副作用が少なく」「合成しやすい」といった条件を満たす化合物を自動で生成することを目指します.
まだまだ発展途上の技術ではありますが, 夢があると思いませんか?
Junction Tree VAE
JT-VAE (junction tree variational autoencoder) [2]は2018年のICMLで発表されたモデルで, 分子生成のベンチマークとなっています.
手法の詳細には踏み込まず, 論文から代表的な図を借りて簡単に説明します.

JT-VAEは, 分子をグラフと木の2通りの表現方法で扱うところが特徴的なモデルです.
原子をノード, 結合をエッジと捉えることで分子はグラフ表現で表すことができますが, 環などの部分構造をひとまとめのノードとすることで木表現 (junction tree) で表すこともできます. グラフと木のそれぞれに対してencoderとdecoderを設けたのがJT-VAEです.
潜在変数$z$がグラフと木で2つ($z_G, z_T$)あることに注意してください.
グラフ生成
それでは, JT-VAEを使って分子グラフの生成を試してみましょう.
コードはこちらのノートブックでもご覧になれます.
準備
v0.3.1ではJTNNDatasetやJTNNCollatorが実装されていませんが, exampleの中にあるものを流用できます.
datautils.py (リンク有)をsite-packages内のdgl/model/chem/jtnn/にコピーして, dgl/model/chem/jtnn/__init__.pyにfrom .datautils import JTNNDataset, JTNNCollatorを追記してください.
前処理など
まずはライブラリをインポートします.
import dgl
from dgl import model_zoo
from dgl.model_zoo.chem.jtnn import JTNNDataset, cuda, JTNNCollator
import rdkit
from rdkit import Chem
from rdkit.Chem import Draw, MolFromSmiles, MolToSmiles
import torch
from torch.utils.data import DataLoader, Subset
次はデータの前処理です. 自分で書いてもよかったのですが, DGLのJTNNDatasetでやってくれるみたいなのでお任せしました.
dataset = JTNNDataset(data="test", vocab="vocab", training=False)
dataset.training = False
*初めて使う場合はvocab.txtが無いというエラーが出ると思います. その場合はdgl/examples/pytorch/model_zoo/chem/generative_models/jtnn/train.pyを実行すると必要な場所にダウンロードされます. 参考: https://github.com/dmlc/dgl/issues/819
先程読まれたDatasetはDGLで用意されたテストデータなので, 自分が使いたいものに置き換えましょう.
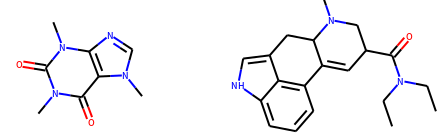
ここではカフェインとリゼルグ酸ジエチルアミドの間の補間をすることにします.
dataset.data = ['CN1C=NC2=C1C(=O)N(C(=O)N2C)C', 'CCN(CC)C(=O)C1CN(C2CC3=CNC4=CC=CC(=C34)C2=C1)C']
ここで入力した文字はSMILESと呼ばれる文字列表現で, PubChemなどで調べることができます.
次はデータのバッチ化ですが, 上手い方法がわからなかったので, (冗長になってしまいますが)Dataloaderを利用します.
def worker_init_fn(id_):
lg = rdkit.RDLogger.logger()
lg.setLevel(rdkit.RDLogger.CRITICAL)
worker_init_fn(None)
dataset.training = False
dataloader = DataLoader(
Subset(dataset, [0,1]),
batch_size=1,
shuffle=False,
num_workers=0,
collate_fn=JTNNCollator(dataset.vocab, False),
drop_last=True,
worker_init_fn=worker_init_fn)
これで準備は完了です. 一旦, datasetに入れたデータを可視化してみましょう.
Draw.MolsToGridImage([MolFromSmiles(s) for s in dataset.data], molsPerRow=4,subImgSize=(250,150))
モデル読み込み
次に, JT-VAEの学習済みモデルを読み込みます.
model = model_zoo.chem.load_pretrained('JTNN_ZINC')
model = cuda(model)
このモデルはVAEの元論文 [3]のMNISTで言うところのこのようなデータ分布を学習しているはずです.

化合物でも, このようにカフェインとリゼルグ酸ジエチルアミドの間を連続的につなぐことはできるでしょうか?
潜在空間での補間
まずはカフェインとリゼルグ酸ジエチルアミドに対応する潜在変数$z_{start}, z_{goal}$を求めます.
tree_vec[0]とmol_vec[0]がカフェイン, tree_vec[1]とmol_vec[1]がリゼルグ酸ジエチルアミドの潜在変数です.
tree_vecs, mol_vecs = [], []
for batch in dataloader:
model.move_to_cuda(batch)
_, tree_vec, mol_vec = model.encode(batch)
tree_vec, mol_vec, _, _ = model.sample(tree_vec, mol_vec) # reparam. trick
tree_vecs.append(tree_vec)
mol_vecs.append(mol_vec)
始点と終点が決まったので, 線分を分割した点を取り, 順にdecodeしていきます. decodeの出力はSMILESです.
# 潜在空間でカフェインからリゼルグ酸ジエチルアミドへ向かうベクトル
tree_diff = tree_vecs[1] - tree_vecs[0]
mol_diff = mol_vecs[1] - mol_vecs[0]
smiles = []
num_mols = 100 # 端点を含めて100個生成
tree_st, mol_st = tree_vecs[0], mol_vecs[0]
# 分割した点を順にdecodeしていく
for i in tqdm(range(num_mols)):
s = model.decode(tree_st+tree_diff/(num_mols-1)*i, mol_st+mol_diff/(num_mols-1)*i)
smiles.append(s)
最後に, 生成した100個の分子を順番に表示します. 結合のルールを守っていないなどでNoneが含まれることがあるので, これらを除外して表示します.
mols = []
for s in smiles:
if s is None:
continue
mol = MolFromSmiles(s)
if mol is not None:
mols.append(mol)
Draw.MolsToGridImage(mols, molsPerRow=4, subImgSize=(250,150))
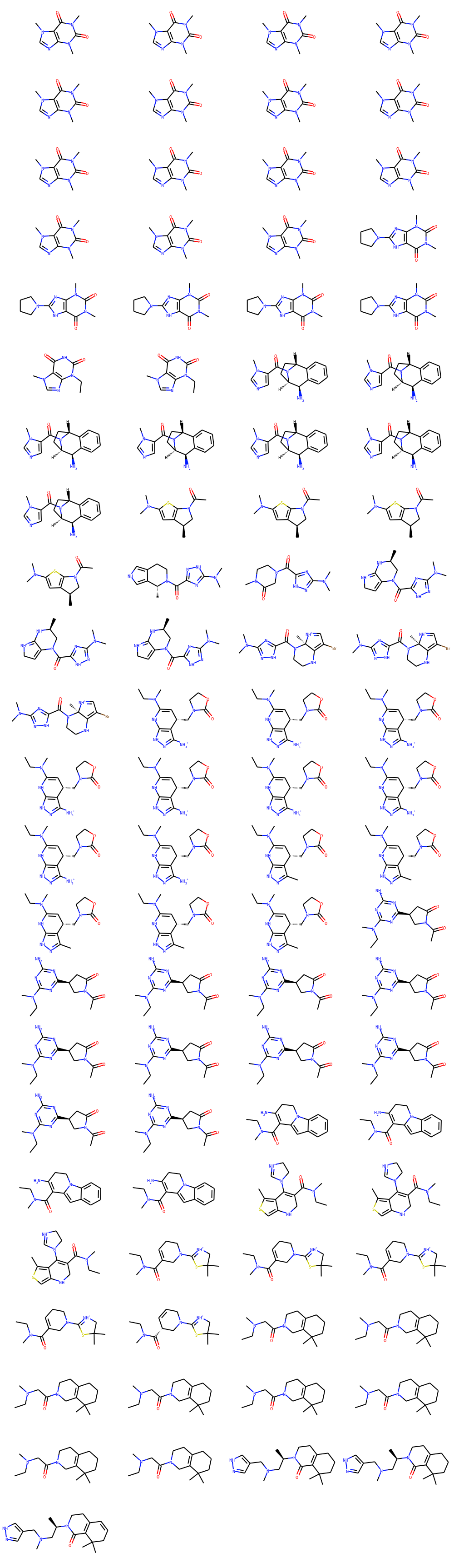
同じ分子が連続して出力されたり, 補間が滑らかでなかったりと, やや微妙な結果ですね.
終点もリゼルグ酸ジエチルアミドに戻っていません…
おわりに
本記事では, DGLの新機能である学習済みJT-VAEを使って分子グラフの補間を試してみました.
今回は微妙な結果でしたが, 最近活発に研究されているテーマなので, 発展が楽しみですね.
このテーマに興味を持ったという方は, 7月に出たサーベイ [4] を読んでみるとおもしろいかもしれません.
参考文献
[1] Rafael Gómez-Bombarelli et al. "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules." ACS Central Science. 2018.
[2] Wengong Jin et al. "Junction Tree Variational Autoencoder for Molecular Graph Generation." ICML. 2018.
[3] Diederik P Kingma & Max Welling. "Auto-Encoding Variational Bayes." ICLR. 2014.
[4] Daniel Schwalbe-Koda et al. "Generative Models for Automatic Chemical Design." 2019.