概要
- EnrichrからGene set libraryをダウンロードしてくる。
- RのパッケージClusterProfilerで、ダウンロードしてきたライブラリを元にエンリッチメント解析を行う。
- 解析結果のテキストと図を出力する。図の幅は自動調整する。
- 計算に時間がかかるので並列計算を行う。
背景
RNA-seq等で興味のある遺伝子リストの特徴を簡単に調べるには、Enrichr、Targetmine、metascapeなどのウェブツールに自分の遺伝子リストをコピペすれば良いかと思います。特にEnrichrはGene Ontologyだけでなく、GEOに登録された各種シークエンス・マイクロアレイデータからNIHグラントを取った研究者の情報、最近ではCOVID-19に関する情報など多様なデータを用いたエンリッチメント解析が行えるのが強みです。
しかしながら、metascape以外の2者は単一の遺伝子リストしか入力に取れない?ため、複数条件の遺伝子リストを比較する作業が煩雑になります。
そこで活躍するのがRのパッケージClusterProfilerです。compareCluster()関数によって複数グループのエンリッチメントを簡単に図示することが出来ます。
図例
そこで本記事では、Enrichrで使用されているデータセットをダウンロードしてきてclusterProfilerで解析する手順をまとめることとしました。
初手の解析として思考停止で実行し、得られた結果を精査することで次の1手を考えるのに有用かと思います。
(IPAなど有償のソフトが使えればそちらの方が良いかと思いますが)
1. EnrichrからGene set libraryをダウンロードする。
- https://maayanlab.cloud/Enrichr/#stats から自分の興味のあるGene set libraryをダウンロードし、同じディレクトリにまとめます。
- ダウンロードしたテキストファイルの拡張子を.txtから.gmtに変更します。この操作は、clusterProfilerで読み込むために必要となります。
2. ClusterProfilerのダウンロード
clusterProfilerはBioconductorから提供されています。
Bioconductorが入っていない人はBioconductorからインストールします。
# install Bioconductor if you don't yet
install.packages("BiocManager")
BiocManager::install(version = "3.11") # depends on R version
# install clusterProfiler
BiocManager::install("clusterProfiler")
3. エンリッチメント解析の実行
解析対象の遺伝子群を遺伝子名のベクトルをリスト形式で用意します。
Enrichrのデータ入力はGene nameなので、clusterProfilerで通常用いるentrez IDのリストではないことに留意してください。
# prepare gene set: a list of "Gene Name” vectors
Sample <- list(
Group1 = vector_of_gene_name1, #e.g., c("EGFR", "TP53", ...)
Group2 = vector_of_gene_name2,
Group3 = vector_of_gene_name3,
Group4 = vector_of_gene_name4
)
次に、Enrichrからダウンロードしてきたデータセットを読み込みます。
# import gene set libraries
mDIR <- "/Dir_to_gmt_files/"
gmt_files <- paste0(mDIR, list.files(mDIR, pattern = ".gmt"))
続いて、データセットのファイルパスを入力としてエンリッチメント解析を行う関数を用意します。
ソースの後に簡単な説明を載せています。
EnrichmentAnalysis <- function(gmt_file, genelist_file, DIR) {
# gmt_file: an item in gmt_files
# genelist_file: gene list of interest
# DIR: directory to export csv and pdf files
# import gmt file
enrichr_gmt <- read.gmt(gmt_file)
# get gmt file name
gmt_file_strip <- str_split(gmt_file, "/", simplify = T)
gmt_file_name <- str_split(gmt_file_strip[length(gmt_file_strip)], "\\.", simplify = T)[1]
print(paste0("calc enrichment with ", gmt_file_name, " ..."))
# calc. enrichment score by culsterProfiler
# enricher() is a function for hypergeometric test
# see http://yulab-smu.top/clusterProfiler-book/chapter3.html
CPfile <- compareCluster(geneCluster = genelist_file, fun='enricher', TERM2GENE=enrichr_gmt)
# export graphs showCategory = 5 or 10
for (j in c(5,10)) {
w <- myWidthAlgorithm(CPfile, j)
g <- dotplot(CPfile, showCategory=j) + ggtitle(gmt_file_name)
ggsave(plot=g, filename=paste0(DIR, "/", gmt_file_name, "_show_", j, ".pdf"),
width=w, h=6)
}
# export table
write_csv(as.data.frame(CPfile), paste0(DIR, "/", gmt_file_name,".csv"))
}
[説明]はじめに、read.gmt()でgmtファイルを読み込みます。
読み込み前のgmtファイル(Enrichrからダウンロードしたtxtファイル)を見ると、1列目にエンリッチメントターム、1列あけて3列目以降に対応するgene nameが並んでいます。
| column 1 | 2 | 3 | 4 | 5 | ... |
|---|---|---|---|---|---|
| ABC transporters | ABCA2 | ABCC4 | |||
| AMPK signaling pathway | G6PC | RAB2A | PPP2R1A | ... | |
| ... | ... |
read.gmt()によって読み込まれたファイルは、1列目にエンリッチメントターム(ont)、2列目にgene name(gene)となるtidyなデータフレームに変換されます。
| ont | gene |
|---|---|
| ABC transporters | ABCA2 |
| ABC transporters | ABCC4 |
| AMPK signaling pathway | G6PC |
| ... | ... |
compareCluster()は、gene setとエンリッチメントに使用する関数を引数に取ります。使用する関数をfun='enrichGO'などとすることで、GO解析など用意された各種解析を行うことが出来ますが、今回は、カスタムgene set libraryから超幾何分布検定を行うenricherを指定します。続いて、enricherの引数として、TERM2GENEにread.gmt()で読み込んだgene list libraryのデータフレームを指定します。
最後に得られた結果から、ドットプロット及びcsvファイルを出力します。
作図に使用しているmyWidthAlgorithm()は、解析結果からエンリッチメントタームの長さと遺伝子グループの数を参照して、出力pdfの幅を決定します。幅を可変にしておかないと、エンリッチメントタームが長い場合にドットプロットが潰れて見えなくなるためです。
myWidthAlgorithm <- function(CPfile, n) {
df <- CPfile@compareClusterResult
df_show <- df %>% group_by(Cluster) %>% top_n(-qvalue, n = n)
maxnchar <- max(nchar(df_show$ID))
nclu <- df$Cluster %>% unique() %>% length()
return(nclu*1.5 + maxnchar/10)
}
最後に、全てのgmtファイルについてEnrichmentAnalysis()を実行します。compareCluster()は有意なエンリッチメントタームがないとエラーを吐いてfor loopが止まってしまうので、tryChatch()でエラーが出た場合はスキップするようにします(参考)。
なお、compareCluster()はそこそこ計算時間を要するため、このままシングルコアで計算すると時間がかかります。
DIR <- "clusterProfiler"
dir.create(DIR)
start.time <- proc.time() # start time
for (i in 1:length(gmt_files)){
skip_to_next <- FALSE
gmt_file <- gmt_files[i]
tryCatch(EnrichmentAnalysis(gmt_file, Sample, DIR),
error = function(e) {skip_to_next <<- TRUE})
if(skip_to_next) { next }
}
end.time <- proc.time() # finish time
(end.time-start.time) # passed time
数百遺伝子x4グループの遺伝子リストに対して、131のgene set library(gmtファイル)のエンリッチメント解析をシングルコアで行った結果、約45分かかりました(実行環境: iMac-Pro 2017 3 GHz Intel Xeon W)。遺伝子リストの要素数やグループ数が多い場合、さらに時間がかかると予想されます。
ユーザ システム 経過
2688.542 60.520 2749.154
そこで上記の計算をforeachおよびdoSNOWを用いて並列化します。
# install.packages(c("foreach", "doSNOW")) # if you don't install yet
library(foreach)
library(doParallel) #use detectCores()
library(doSNOW)
# prepare parallel computing
# setup parallel backend to use many processors
cores=detectCores()
cl <- makeCluster(cores[1]-1) # not to overload your computer
invisible(clusterEvalQ(cl, {library(clusterProfiler)})) # to load required library in each core
registerDoSNOW(cl)
start.time <- proc.time()
foreach(i=1:length(gmt_files3)) %dopar% { # single core: %do% {
skip_to_next <- FALSE
gmt_file <- gmt_files3[i]
tryCatch(EnrichmentAnalysis(gmt_file, Sample, DIR),
error = function(e) {skip_to_next <<- TRUE})
if(skip_to_next) { next }
}
end.time <- proc.time()
(end.time-start.time)
stopCluster(cl) # stop parallel computing
同様に131のgmtファイルの解析を9コアで行った計算時間は約6分となり、約1/8程度に短縮できました。
ユーザ システム 経過
3.798 2.911 359.671
結果の一例
結果の一例として、せっかくなので変わり種を置いておきます。
下の図は、NIHグラント受給者の論文で出現した遺伝子リストのgene libraryを使って、エンリッチメント解析を行った結果です。(gene libraryに関する説明: The release of modEnrichr and new libraries for genes studied by NIH-funded PIs)
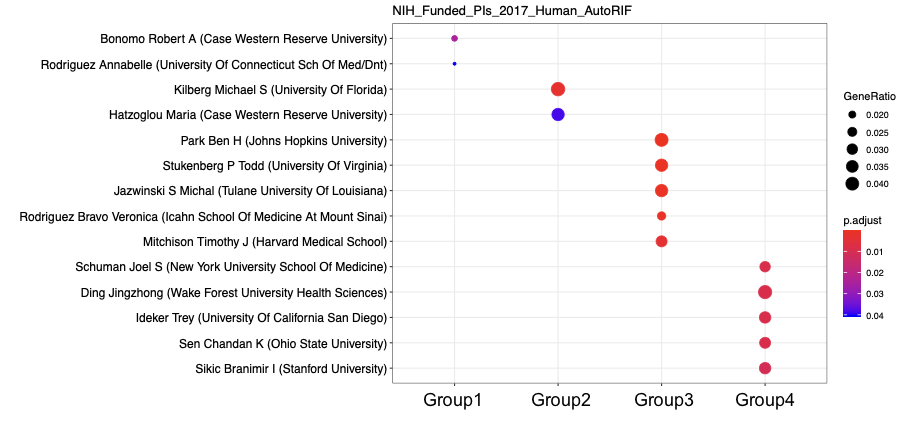
出てきた研究者のウェブサイトや論文が自身の研究に役に立つことがもしかしたらあるかもしれません。