volcano plotとは、RNA-seq解析をした際に発現比較解析の結果を示すグラフの一つです。横軸に発現比(log2)、縦軸にはp値(-log10)をとります。
R言語のplot関数を用いてvolcano plotを描く方法を以前記事にしましたが、今回はシミュレーションデータを作成し、そこからggplot2を用いてvolcano plotを描く方法を備忘録としてまとめたいと思います。
前回の記事はこちらから
なお、筆者の用いたR studioはVersion 1.4.1106、Rはversion 4.0.5を用いました。ライブラリのバージョンは以下のスクリプト内に記載しています。
シミュレーションデータの作成
まず、volcano plotを描くためのシミュレーションデータを作成していきます。
RのTCC packageを用いてデータを作成しました。
TCC packageについては、Sunらの論文を参照してください(ページ最下部に記してます)。
シミュレーションデータの作成は以下のURLを参考にしました。
以下のスクリプトを用いて、シミュレーションデータをcsvファイルに書き出します。
今回は、biological replicateを3、2群間比較とし、iDEGES/edgeRで発現変動解析を行いました。
library(TCC)
packageVersion("TCC")
packageVersion("edgeR")
count <- simulateReadCounts(Ngene = 10000, PDEG = 0.20, DEG.assign = NULL,
DEG.foldchange = NULL, replicates = NULL, group = NULL,
fc.matrix = NULL)
in_f <- count$count
out_f <- "TCC_simulation_data.csv"
param_A <- 3
param_B <- 3
param1 <- 3
param_DEmethod <- "edger"
param_FDR <- 0.05
data <- as.matrix(in_f)
data.cl <- c(rep(1, param_A), rep(2, param_B))
data.cl
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc,iteration=param1)
tcc <- estimateDE(tcc, test.method=param_DEmethod, FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.csv(tmp, out_f, append=F, quote=T, row.names=F)
volcano plotの作成
さて、ここからvolcano plotを描いていきます。
まずは、必要なパッケージの読み込みとデータの読み込みを行います。
library(ggplot2)
library(dplyr)
packageVersion("ggplot2")
packageVersion("dplyr")
countdata <- read.csv("TCC_simulation_data.csv")
head(countdata)
次に、volcano plotを描いた際に、発現が上昇した遺伝子、低下した遺伝子それぞれのプロットを色付けするための条件を記述し、新たな変数としてcountdataに追加します。
countdata <- mutate(countdata, thcolor = ifelse(p.value >= 0.05 , 0,
ifelse(m.value >= 1, 1,
ifelse(m.value <= -1, 2,0))))
p値が0.05未満で、fold change(m.valueのこと)が1以上の時は1、fold changeが-1以下の時は2、それ以外は0を指定しています。
いよいよグラフの作成です。
ggplot() +
geom_point(aes(x = countdata$m.value, y = -log10(countdata$p.value), color = as.factor(countdata$thcolor)), size = 0.1) +
labs(x = "log2 (Fold Change)", y = "-log10 (p-value)") +
scale_color_manual(values = c("black", "red", "blue")) +
geom_hline(yintercept = -log10(0.05), size = 0.2, color = "dark green") +
geom_vline(xintercept = -1, size = 0.2, color = "dark green") +
geom_vline(xintercept = 1, size = 0.2, color = "dark green") +
theme(legend.position = "none", panel.grid = element_blank())
fold changeが1以上の遺伝子を赤、-1以下のものを青、それ以外を黒色でプロットしました。
また、閾値を分かりやすくするため、深緑色の線を引きました。
volcano plotはp値を対数変換する必要があるので、-log10()で変換しました。
以上のスクリプトを実行し描いたグラフは以下のようになります。
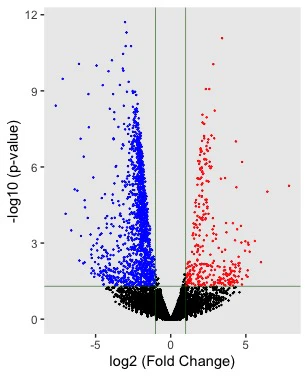
発現変動遺伝子が分かりやすく可視化されました。
plot関数よりもggplot2の方がこだわれば、よりきれいなグラフが描くことができるかもしれません。(少なくとも私自身はplot関数で描くのと、どちらが良いか分かりませんが。。。)
最後に、本記事で用いたスクリプトを実行した結果を以下に示します。
> library(TCC)
~読み込みが長いので割愛~
> packageVersion("TCC")
[1] ‘1.30.0’
> packageVersion("edgeR")
[1] ‘3.32.1’
> count <- simulateReadCounts(Ngene = 10000, PDEG = 0.20, DEG.assign = NULL,
+ DEG.foldchange = NULL, replicates = NULL, group = NULL,
+ fc.matrix = NULL)
TCC::INFO: Generating simulation data under NB distribution ...
TCC::INFO: (genesizes : 10000 )
TCC::INFO: (replicates : 3, 3 )
TCC::INFO: (PDEG : 0.18, 0.02 )
> in_f <- count$count
> out_f <- "TCC_simulation_data.csv"
> param_A <- 3
> param_B <- 3
> param1 <- 3
> param_DEmethod <- "edger"
> param_FDR <- 0.05
> data <- as.matrix(in_f)
> data.cl <- c(rep(1, param_A), rep(2, param_B))
> data.cl
[1] 1 1 1 2 2 2
> tcc <- new("TCC", data, data.cl)
> tcc <- calcNormFactors(tcc,iteration=param1)
TCC::INFO: Calculating normalization factors using DEGES
TCC::INFO: (iDEGES pipeline : tmm - [ edger - tmm ] X 3 )
TCC::INFO: Done.
> tcc <- estimateDE(tcc, test.method=param_DEmethod, FDR=param_FDR)
TCC::INFO: Identifying DE genes using edger ...
TCC::INFO: Done.
> result <- getResult(tcc, sort=FALSE)
> tmp <- cbind(rownames(tcc$count), tcc$count, result)
> write.csv(tmp, out_f, append=F, quote=T, row.names=F)
警告メッセージ:
write.csv(tmp, out_f, append = F, quote = T, row.names = F) で:
'append' への変更の試みは無視されました
> library(ggplot2)
> library(dplyr)
次のパッケージを付け加えます: ‘dplyr’
以下のオブジェクトは ‘package:baySeq’ からマスクされています:
groups
以下のオブジェクトは ‘package:Biobase’ からマスクされています:
combine
以下のオブジェクトは ‘package:matrixStats’ からマスクされています:
count
以下のオブジェクトは ‘package:GenomicRanges’ からマスクされています:
intersect, setdiff, union
以下のオブジェクトは ‘package:GenomeInfoDb’ からマスクされています:
intersect
以下のオブジェクトは ‘package:IRanges’ からマスクされています:
collapse, desc, intersect, setdiff, slice, union
以下のオブジェクトは ‘package:S4Vectors’ からマスクされています:
first, intersect, rename, setdiff, setequal, union
以下のオブジェクトは ‘package:BiocGenerics’ からマスクされています:
combine, intersect, setdiff, union
以下のオブジェクトは ‘package:stats’ からマスクされています:
filter, lag
以下のオブジェクトは ‘package:base’ からマスクされています:
intersect, setdiff, setequal, union
>
> packageVersion("ggplot2")
[1] ‘3.3.3’
> packageVersion("dplyr")
[1] ‘1.0.5’
> countdata <- read.csv("TCC_simulation_data.csv")
> head(countdata)
rownames.tcc.count. G1_rep1 G1_rep2 G1_rep3 G2_rep1 G2_rep2 G2_rep3 gene_id a.value m.value
1 gene_1 50 27 32 10 13 10 gene_1 4.320660 -1.688783
2 gene_2 23 55 28 8 4 0 gene_2 3.571005 -3.115365
3 gene_3 246 256 158 106 54 74 gene_3 7.032787 -1.463880
4 gene_4 87 47 68 14 21 10 gene_4 4.989496 -2.131754
5 gene_5 270 137 235 50 46 115 gene_5 6.938669 -1.569962
6 gene_6 177 151 116 50 44 47 gene_6 6.381648 -1.621381
p.value q.value rank estimatedDEG
1 0.0105380207 0.078700677 1339 0
2 0.0008475635 0.008552609 991 1
3 0.0008382452 0.008492859 987 1
4 0.0002320392 0.002726665 851 1
5 0.0013314925 0.012827481 1038 1
6 0.0001367345 0.001700678 804 1
> countdata <- mutate(countdata, thcolor = ifelse(p.value >= 0.05 , 0,
+ ifelse(m.value >= 1, 1,
+ ifelse(m.value <= -1, 2,0))))
>
> ggplot() +
+ geom_point(aes(x = countdata$m.value, y = -log10(countdata$p.value), color = as.factor(countdata$thcolor)), size = 0.1) +
+ labs(x = "log2 (Fold Change)", y = "-log10 (p-value)") +
+ scale_color_manual(values = c("black", "red", "blue")) +
+ geom_hline(yintercept = -log10(0.05), size = 0.2, color = "dark green") +
+ geom_vline(xintercept = -1, size = 0.2, color = "dark green") +
+ geom_vline(xintercept = 1, size = 0.2, color = "dark green") +
+ theme(legend.position = "none", panel.grid = element_blank())
reference
Jianqiang Sun, Tomoaki Nishiyama, Kentaro Shimizu, Koji Kadota. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics. 2013, 14(219) doi: 10.1186/1471-2105-14-219.