2024-02-28 追記:
「大型DNAウイルスゲノムの解析 (2) アノテーション編」を公開する予定です。
内容については、Kawato et al. (2023) "Integrase-associated niche differentiation of endogenous large DNA viruses in crustaceans"のSupplementary Filesをご覧ください。
大型DNAウイルスゲノムの解析
Oxford Nanopore TechnologiesロングリードとIlluminaショートリードでシーケンシングした、未分離の (=宿主配列が混入した)大型DNAウイルスのゲノム配列を解析する方法について記録しておく。
今回用いるデータはヒラメに感染する大型DNAウイルスのリンホシスチス病ウイルス。
病魚腫瘍から直接DNA抽出したため、宿主であるヒラメの配列が混じっている。
Lymphocystis disease virus 2 LCDV-JP_Oita_2018 DNA, complete genome

Genome Sequence of Lymphocystis Disease Virus 2 LCDV-JP_Oita_2018, Isolated from a Diseased Japanese Flounder (Paralichthys olivaceus) in Japan
- Unix環境 (WSL含む) でのCUI操作にある程度慣れていることを想定する
-
cd,mkdir,wget等のコマンドが使えること -
絶対パス/相対パスを扱えること
-
.がcurrent directory (現在地)、..が親ディレクトリを意味すること -
コマンド右側の
#がコメントアウトを意味すること
など。
-
目次
必要ソフトのインストール
必要なソフトをcondaあるいはmamba経由でインストールする。
特定のステップでしか使わないand/or依存関係が複雑なソフトは、個別に環境を用意するのがよい。
# Replace "conda" with "mamba" if using mamba
conda install -y -c bioconda sra-tools
conda install -y -c bioconda seqkit
conda install -y -c bioconda samtools
conda install -y -c bioconda minimap2
conda install -y -c bioconda gffread
# Recommend createing dedicated environments for those programs that will be used only at specific stages and/or have complicated dependencies
conda create -n flye-2.9.1 -y -c bioconda flye=2.9.1
conda create -n masurca-4.0.9 -y -c bioconda masurca=4.0.9
conda create -n prodigal-2.6.3 -y -c bioconda prodigal=2.6.3
conda create -n gff3toddbj-0.4.0 -y -c bioconda gff3toddbj=0.4.0
WSL2の場合、画面をポチポチして動かす (GUI) ソフトを動かすのがめんどくさい(かった?)。
そこで、以下のソフトはWindowsバージョンを別途ダウンロードし、Windowsで直に走らせていただきたい。
Bandage
Integrative Genomics Viewer (IGV)
作業ディレクトリを作る
今回は/path/to/study/(実際にやる際は/home/kawato/とかだったりする)の直下に2023-01-01_LCDV (つまり、YYYY-MM-DD_プロジェクト名) というディレクトリを作る。
さらにその直下に
-
reads: 生リードを保存 -
scripts: スクリプトやログ(どちらも実験ノートのようなもの)を保存 -
analysis: 解析結果を保存
の3つのディレクトリを作る。
自分はこうやっているというだけで特に決まりはない。各自好きにしてほしい。
cd /path/to/study/
mkdir 2023-01-01_LCDV
cd 2023-01-01_LCDV
mkdir reads scripts analysis
元データの入手 (wget)
今回は練習なので、公共データベース上にアップロードされている(=ネットに落ちてる)データを利用する。
自分シーケンスしたデータを用いる場合はこのステップは飛ばす。
FASTQファイルをダウンロードする場合
FASTQファイルが直接ダウンロード可能な場合はそれが手っ取り早い。ほとんどの場合European Nucleotide Archive (ENA)からダウンロードできる。
DDBJから登録されたファイルはDDBJからfastqがダウンロード可能。
ターミナル上でのダウンロードにはwgetコマンドを使う。
cd reads
# Download FASTQ files directly if available at DDBJ or ENA.
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA009/DRA009759/DRX204236/DRR213899_1.fastq.bz2 # Illumina reads, R1
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA009/DRA009759/DRX204236/DRR213899_2.fastq.bz2 # Illumina reads, R2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA009/DRA009759/DRX204237/DRR213900.fastq.bz2 # ONT reads
less DRR213899_1.fastq.bz2 |gzip >DRR213899_1.fastq.gz & # convert to .gz
less DRR213899_2.fastq.bz2 |gzip >DRR213899_2.fastq.gz & # convert to .gz
less DRR213900.fastq.bz2 |gzip >DRR213900.fastq.gz & # convert to .gz
# Most SRA entries are available at ENA in FASTQ format
# wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/DRR213/DRR213899/DRR213899_1.fastq.gz # Illumina reads, R1 (ENA)
# wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/DRR213/DRR213899/DRR213899_2.fastq.gz # Illumina reads, R2 (ENA)
# wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/DRR213/DRR213900/DRR213900_1.fastq.gz # ONT reads (ENA)
SRA形式ファイルをダウンロードする場合
FASTQがダウンロードできない場合、SRA形式のファイルをダウンロードしてあとからFASTQに変換する。
NCBIは通常SRAしかDLできない。
# Download SRA files if FASTQ files are unavailable
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/DRR213899/DRR213899
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/DRR213900/DRR213900
SRAファイルの変換 (sra-tools)
fasterq-dump DRR213899
spots read : 3,682,911
reads read : 7,365,822
reads written : 7,365,822
gzip DRR213899_1.fastq # .gz に圧縮
gzip DRR213899_2.fastq
fasterq-dump DRR213900
spots read : 97,124
reads read : 97,124
reads written : 97,124
gzip DRR213900.fastq
リードの前処理 (QC)
シーケンサーから出力されたそのままのデータ(生データ)には、品質の低い配列が混じっていることが多い。そのまま解析に用いると結果に悪影響を及ぼす可能性があるため、事前に取り除く(quality control: QC)。
シェルスクリプトを使いましょう
普通の実験 (ウェットラボ)で実験ノートをとるのと同様に、解析(ドライラボ)でもやったことを記録しておきたい。
そこで、解析コマンドをシェルスクリプトという形式で書き記し、コンピューターに実行させることで解析ログを記録する。YYYY-MM-DD_(analysis).shのような名前で保存している。
IlluminaリードのQC (Fastp)
クオリティ が低かったりアダプタ配列が混じっていたりする場合があるのでそれらを取り除く。
#!/bin/bash
# >>>>>>
# 2023-01-01_DRR213899_fastp.sh
# >>>>>>
source ~/miniconda3/etc/profile.d/conda.sh # シェルスクリプトでcondaを使うときのおまじない。
#source ~/anaconda3/etc/profile.d/conda.sh # シェルスクリプトでcondaを使うときのおまじない。anaconda3のばあいはこっち
# Create/goto WDIR
WHOME=/path/to/study/2023-01-01_LCDV/reads
cd ${WHOME}
# Set VARs
NUMTH=6
conda activate fastp-0.23.2
RUN=DRR213899
fastp \
-i ${RUN}_1.fastq.gz \
-I ${RUN}_2.fastq.gz \
-o ${RUN}_1_trim.fq.gz \
-O ${RUN}_2_trim.fq.gz \
--html ${RUN}_fastp.html \
--thread ${NUMTH} \
nohupコマンドでジョブを「投げる」(ジョブスケジューラではないのだが)。
cd ../scripts/ # reads からscriptsへ移動
nohup ./2023-01-01_DRR213899_fastp.sh 2>2023-01-01_DRR213899_fastp.err &
[2] 442 # 毎回数字は変わる
[2]+ Done nohup ./2023-01-01_DRR213899_fastp.sh 2> 2023-01-01_DRR213899_fastp.err
less 2023-01-01_DRR213899_fastp.err
nohup: ignoring input and appending output to 'nohup.out'
Read1 before filtering:
total reads: 3682911
total bases: 433382840
Q20 bases: 422090448(97.3944%)
Q30 bases: 419067756(96.6969%)
Read2 before filtering:
total reads: 3682911
total bases: 434053421
Q20 bases: 409482405(94.3392%)
Q30 bases: 404652168(93.2264%)
Read1 after filtering:
total reads: 3659510
total bases: 431953247
Q20 bases: 421626778(97.6094%)
Q30 bases: 418653103(96.9209%)
Read2 after filtering:
total reads: 3659510
total bases: 431818313
Q20 bases: 408830407(94.6765%)
Q30 bases: 404149409(93.5925%)
Filtering result:
reads passed filter: 7319020
reads failed due to low quality: 46802
reads failed due to too many N: 0
...
ONTリードのQC (SeqKit)
短かすぎる (※) リードを取り除く。クオリティでは特にフィルタリングしていない。
#!/bin/bash
# >>>>>>
# 2023-01-01_DRR213900_filter.sh
# >>>>>>
source ~/miniconda3/etc/profile.d/conda.sh # シェルスクリプトでcondaを使うときのおまじない。
#source ~/anaconda3/etc/profile.d/conda.sh # シェルスクリプトでcondaを使うときのおまじない。anaconda3のばあいはこっち
# Create/goto WDIR
WHOME=/path/to/study/2022-01-01_LCDV/reads
cd ${WHOME}
RUN=DRR213900
seqkit seq -j 4 -m 5000 ${RUN}.fastq.gz -o ${RUN}.up5k.fq.gz &
seqkit seq -j 4 -m 10000 ${RUN}.fastq.gz -o ${RUN}.up10k.fq.gz &
seqkit seq -j 4 -m 20000 ${RUN}.fastq.gz -o ${RUN}.up20k.fq.gz &
wait
seqkit stats ${RUN}.fastq.gz ${RUN}*.fq.gz >${RUN}.stats
nohup ./2023-01-01_DRR213900_filter.sh 2>2023-01-01_DRR213900_filter.err &
[1]+ Done nohup ./2023-01-01_DRR213900_filter.sh 2> 2023-01-01_DRR213900_filter.err
cd ../reads/
less DRR213900.stats
file format type num_seqs sum_len min_len avg_len max_len
DRR213900.fastq.gz FASTQ DNA 97,124 630,612,147 71 6,492.9 184,865
DRR213900.up10k.fq.gz FASTQ DNA 16,541 394,118,967 10,000 23,826.8 184,865
DRR213900.up20k.fq.gz FASTQ DNA 7,400 266,192,623 20,000 35,972 184,865
DRR213900.up5k.fq.gz FASTQ DNA 31,634 500,072,397 5,000 15,808.1 184,865
※ONTのリード長について
一概に言えないが、
- 1-3 kbは明らかに短すぎる。
- 10 kb未満まで切り捨てると、バクテリアゲノムのプラスミドを取りこぼしてしまうかもしれない。
- 5-10 kbのリードを残しても長大な反復領域をまたぎ切れず、アセンブリ結果が不正確になるかもしれない。
- あまりリードの長さにこだわると、今度はカバレッジが不足してしまうかもしれない。
全てのデータに一律に適用可能な基準はない。いろいろ長さの閾値を変えてアセンブリを試してほしい。
De novo assembly (Flye)
ONTリードからもとのゲノム配列を再構築 (アセンブル assemble) する。参照ゲノム等に基づいて (reference-based) ではなく新規に (de novo) 再構築するから de novo assembly.
1回目 (metaFlye)
長さでフィルタリングして残ったONTリードを、メタゲノムモード (--meta) でアセンブルする。5kb, 10kb, 20kbの3つの閾値をとってみる。
#!/bin/bash
# >>>>>>
# 2023-01-01_DRR213900_flye_meta.sh
# >>>>>>
source ~/miniconda3/etc/profile.d/conda.sh
# Create/goto WDIR
WHOME=/path/to/study/2022-01-01_LCDV/analysis/
WDIR=2023-01-01_DRR213900_flye_meta
cd ${WHOME}
mkdir ${WDIR};cd ${WDIR}
# Set VARs
NUMTH=20
RUN=DRR213900
### Flye ###
conda activate flye-2.9.1
for N in 20 10 5 ;do
flye --meta \
--nano-raw ../../reads/${RUN}.up${N}k.fq.gz \
--out-dir flye_up${N}k_meta \
--threads ${NUMTH}
done
conda deactivate
nohup ./2023-01-01_DRR213900_flye_meta.sh 2>2023-01-01_DRR213900_flye_meta.err &
[1] 686
今回は20kb以上の区 (flye_up20k_meta) を採用する。
結果として以下のようなファイルができる。
cd ../analysis/2023-01-01_DRR213900_flye_meta/flye_up20k_meta/
ls -lhrt
total 1.7M
drwxrwxrwx 1 kawato docker 4.0K Jan 2 18:02 00-assembly
drwxrwxrwx 1 kawato docker 4.0K Jan 2 18:05 10-consensus
drwxrwxrwx 1 kawato docker 4.0K Jan 2 18:06 20-repeat
drwxrwxrwx 1 kawato docker 4.0K Jan 2 18:06 30-contigger
drwxrwxrwx 1 kawato docker 4.0K Jan 2 18:09 40-polishing
-rwxrwxrwx 1 kawato docker 93 Jan 2 18:09 params.json
-rwxrwxrwx 1 kawato docker 4.0K Jan 2 18:09 assembly_graph.gv
-rwxrwxrwx 1 kawato docker 685K Jan 2 18:09 assembly_graph.gfa
-rwxrwxrwx 1 kawato docker 698K Jan 2 18:09 assembly.fasta
-rwxrwxrwx 1 kawato docker 514 Jan 2 18:09 assembly_info.txt
-rwxrwxrwx 1 kawato docker 75K Jan 2 18:09 flye.log
使うのは、assembly_graph.gfa(アセンブリグラフ)。
Bandageでassembly_graph.gfaを可視化する。
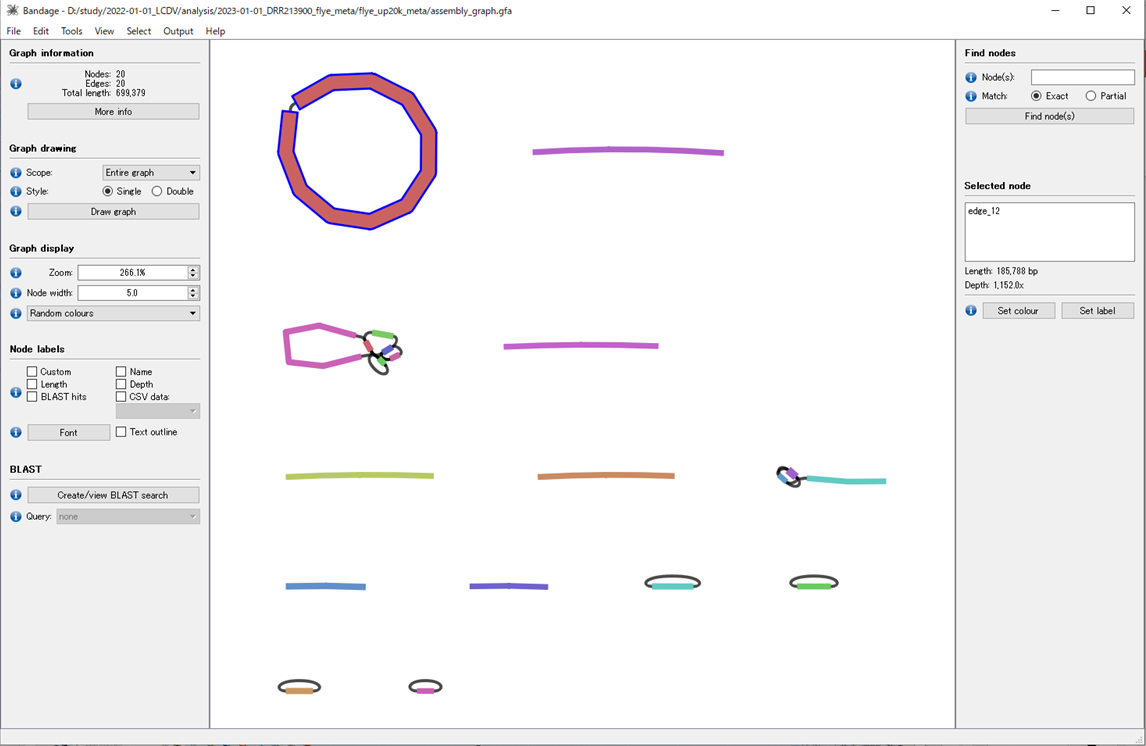
edge_12が目的のウイルス配列。
Save selected node sequences to FASTAで配列を保存。
2回目 (Flye)
ウイルスのドラフト配列に貼り付いたONTリードを通常モードでアセンブルする。
samtools view -q 60も試したがPAFからフィルタリングしたほうがうまくいく(※)。
#!/bin/bash
# >>>>>>
# 2023-01-02_edge_12_flye.sh
# >>>>>>
source ~/miniconda3/etc/profile.d/conda.sh
# Create/goto WDIR
WHOME=/path/to/study/2022-01-01_LCDV/analysis/
WDIR=2023-01-02_edge_12_flye
cd ${WHOME}
mkdir ${WDIR};cd ${WDIR}
# Set VARs
NUMTH=20
N=20
RUN=DRR213900
LONGR=../../reads/${RUN}.up${N}k.fq.gz
(minimap2 -t ${NUMTH} -x map-ont ../2023-01-01_DRR213900_flye_meta/flye_up20k_meta/edge_12.fasta ${LONGR} >${RUN}.up${N}k.paf)2>${RUN}.up${N}k.minimap2.log
awk '{FS="\t"; OFS="\t"};$10/$11 >= 0.7 && $11/$2 >= 0.3 {print $1}' ${RUN}.up${N}k.paf |sort -u > ${RUN}.up${N}k.mapped.list
seqkit grep -f ${RUN}.up${N}k.mapped.list ${LONGR} | gzip >${RUN}.up${N}k.mapped.fq.gz
conda activate flye-2.9.1
flye --nano-raw ${RUN}.up${N}k.mapped.fq.gz --out-dir flye_up${N}k --threads ${NUMTH}
conda deactivate
※mapqはリードがレファレンス上のある領域にユニークにマップされたということを保証するだけで、実際にリード全体の何割がレファレンスに貼り付いたかを考慮していない。極端な例になると、数十bp貼り付いただけでもそれがユニークならマッピングクオリティは高くなる。
Bandageでアセンブリグラフ (assembly_graph.gfa)を可視化する。
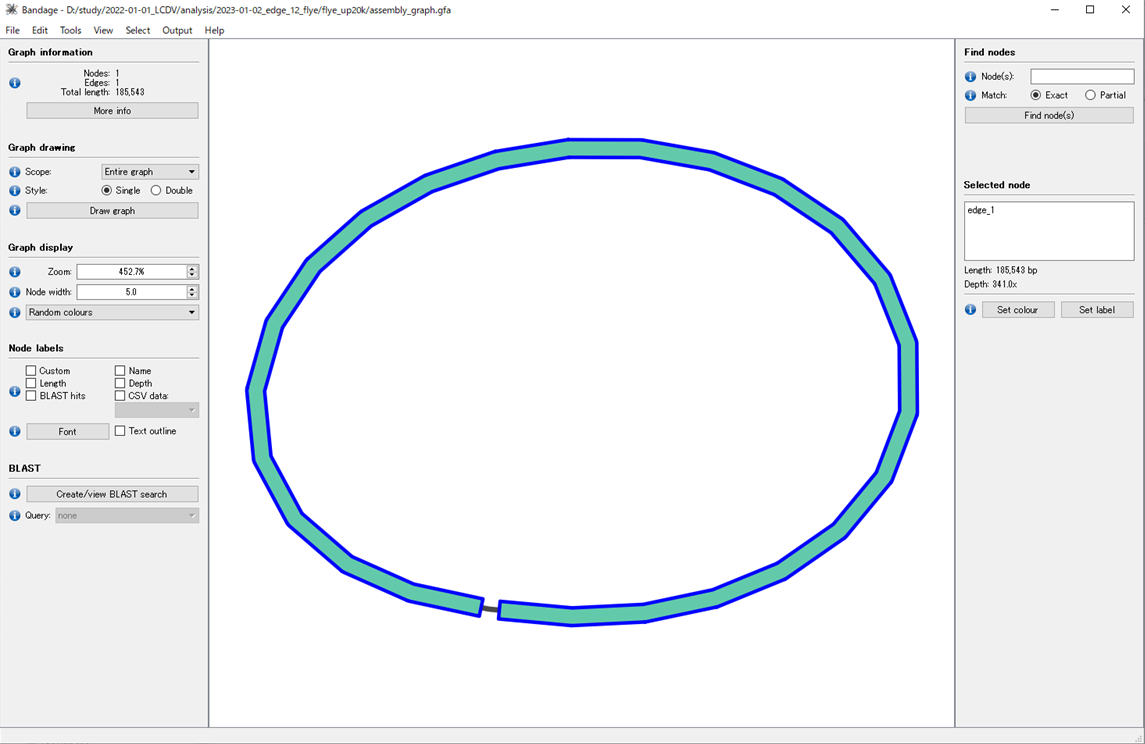
1本の環状ゲノムがアセンブルできた。
エラー修正 (ポリッシング polishing)
ロングリードをアセンブルして得られた配列はエラーを多く含む「粗削り」の状態であるため、正確性に優れるショートリードを利用してエラー修正(ポリッシング polishing) を行う。PacBio HiFiリードやONT V14といった最新のロングリードをアセンブルした場合はpolishingは必要ないかもしれない。
環状ゲノムの場合、複数回ゲノムを回転させてまんべんなく研磨できるようにしている。そこまでこだわる必要はないかもしれないが、おまじないとしてそうしている。
ショートリードを用いたエラー修正 (POLCA)
#!/bin/bash
# >>>>>>
# 2023-01-02_polca.sh
# >>>>>>
source ~/miniconda3/etc/profile.d/conda.sh
# Create/goto WDIR
WHOME=/path/to/study/2022-01-01_LCDV/analysis/
cd ${WHOME}
# Set VARs
NUMTH=20
mkdir 2023-01-02_polca
cd 2023-01-02_polca
R1=../../../reads/DRR213899_1_trim.fq.gz
R2=../../../reads/DRR213899_2_trim.fq.gz
# Round 1
NUM=0
seqkit seq ../2023-01-02_edge_12_flye/flye_up20k/assembly.fasta > polca0.fa
for NUM in 0 1 2 3 4 5 6 7 8 9 ;do
samtools faidx polca${NUM}.fa
mkdir polca${NUM};cd polca${NUM}
conda activate masurca-4.0.9
polca.sh -a ../polca${NUM}.fa -r "${R1} ${R2}" -t 20
cd ..
conda deactivate
seqkit restart -i 50000 polca${NUM}/polca${NUM}.fa.PolcaCorrected.fa >polca$((NUM + 1)).fa
done
nohup ./2023-01-02_polca.sh 2>2023-01-02_polca.err &
1回目POLCAの出力ファイルのひとつpolca1.fa.reportを見てみる。
Substitution Errors: 0
Insertion/Deletion Errors: 0
Assembly Size: 186316
Consensus Quality: 100
Consensus QV: 100.00
Errorsはいずれも0で、Qualityは100。修正する箇所がなかった(=収束した)と判断した。
10回のポリッシングは必ずしも必要ない。たいてい2-3回もやれば変化はなくなる。本サンプルは1回で収束した。むしろ10回やっても収束しない場合、サンプル内多型(混合感染)を疑ったほうがいい。
出力した配列をSnapGene Viewerで可視化する。
ポリッシュ前 (polca0.fa)

ポリッシュ後 (polca1.fa)
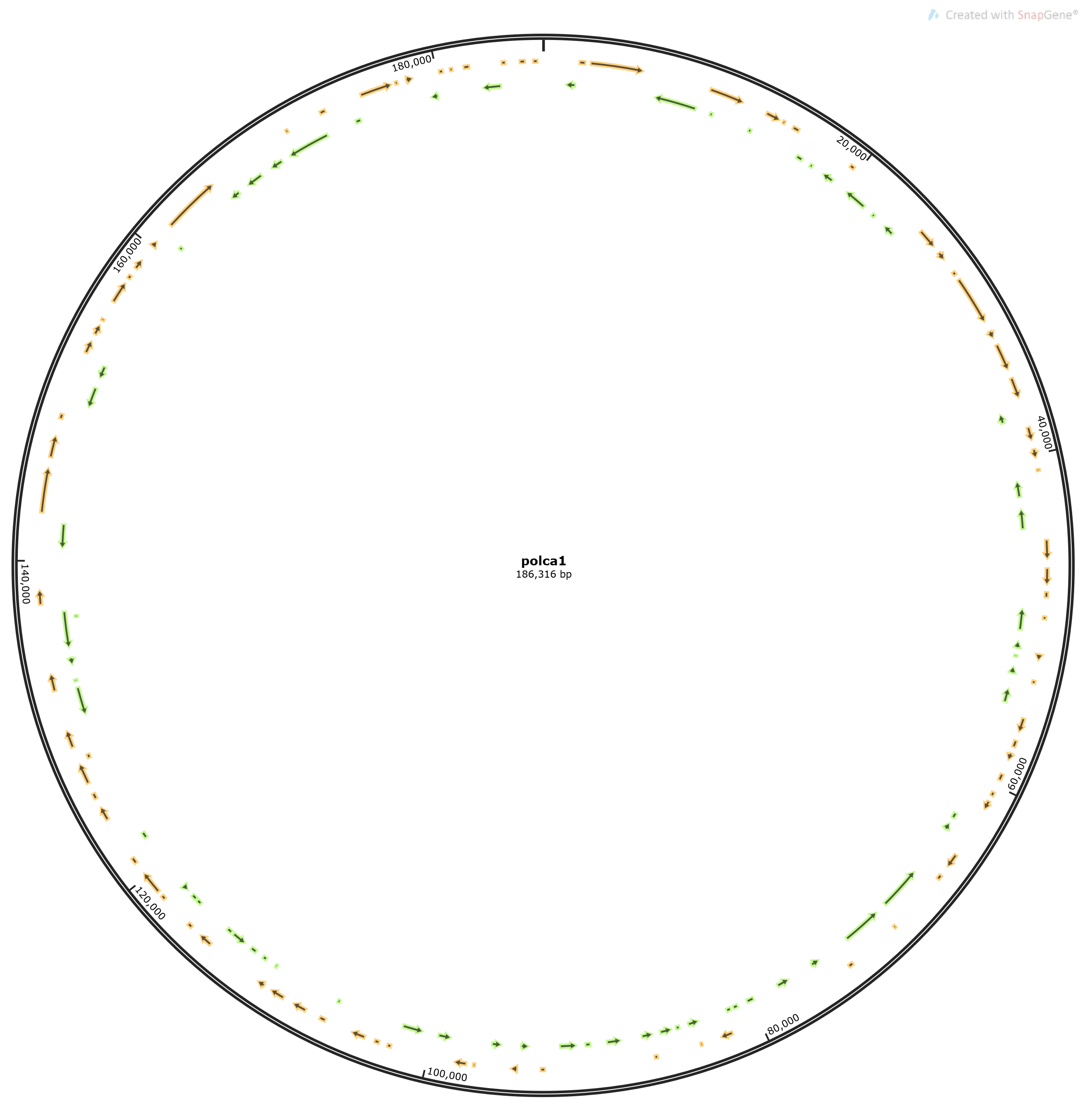
ORF(黄・緑色の矢印)の長さが明らかに違うことがわかる。誤解を恐れず単純に言えば、ORFが長いほうが信頼できる。
これで配列は完成。