シリーズの見出し
1, 環境構築
2, RNA-Seq一連の流れ;トリミング->マッピング->発現量算出->DEG検出->GO解析
レファレンスのある生物種におけるhisat2を用いたマッピング,stringtieを用いたリードカウント,edgeRによるDEG検出、DAVIDでのGO解析、pathway解析
3,de novo アセンブリによる発現量算出
リファレンスのない生物種でde novo asemblyを行って発現定量を行う
2 RNA-Seq一連の流れ
流れ
・テストランデータの取得
・ツールのインストール
・リファレンスデータの取得とインデックスファイルの作成
・トリミング&QC (Trim-galore!)
・マッピング(HISAT2)
・発現定量(SAMtools, StringTie,deepTools)
・csv変換(prepDE.py)
・正規化、DEG検出(edgeR)
・GO解析(DAVID)
参考文献
「RNA-Seqデータ解析 WETラボのための鉄板レシピ Chapter3 転写産物の発現を定量する (1)リファレンスゲノムにマッピングする方法①」
https://bi.biopapyrus.jp/rnaseq/mapping/hista/hisat2-single-rnaseq.html
https://bi.biopapyrus.jp/rnaseq/mapping/bowtie2/bowtie2-signleend.html
リファレンスの入手先
https://jp.support.illumina.com/sequencing/sequencing_software/igenome.html?langsel=/jp/
https://m.ensembl.org/info/data/ftp/index.html
#2.0 マッピングテストラン用データセットの準備
環境
マシン:Macbook Pro (16-inch, 2019) 2.3 GHz 8コアIntel Core i9 メモリ64 GB 2667 MHz DDR4
OS:Mac OS Catalina v10.15.7
requirments:bash(zshでも良いと思う), python3, R, Rstudio
テストランのデータセットはH氏がメダカで取得した生データ(マクロジェンからの返却データ)を使おう
*公開SRAデータを使う場合はSRAからfasta形式への変換やインデックス付けなどの作業が別途いる、これらについては別の記事で解説する予定(21.07.05)
流れ
・テストランデータの取得
・ツールのインストール
・リファレンスデータの取得とインデックスファイルの作成
・トリミング&QC (Trim-galore!)
・マッピング(HISAT2)
・発現定量(SAMtools, StringTie,deepTools)
2.1 テストラン用生データの入手
sample:メダカ肝臓から抽出したtotal mRNA、外注先=Macrogen、2群(n=1)、paired-end、read = 101bp、Library kit = TruSeq stranded mRNA
、Sequencer = Illumina platform
H氏からデータ提供(cab1とcab5の2個体のデータ,2試験区、複製なし)
本来はbiological replicatesはn=3以上が望ましいらしい
cab_liver_1_1.fastq.gz
cab_liver_1_2.fastq.gz
cab_liver_5_1.fastq.gz
cab_liver_5_2.fastq.gz
2.2 ツールのインストール
Trim-galore,bowtie2, HISAT2,SAMtools,StringTie,deepToolsのインストール
conda install -c bioconda trim-galore
conda install -c bioconda hisat2
conda install -c bioconda stringtie
conda install -c bioconda samtools
conda install -c bioconda deeptools
2.3 リファレンスの準備
マッピング先となるリファレンスファイルの準備を行う。全ゲノム配列が決定されている生物に限定される。
EnsembleのFTPダウンロードというサイトへアクセス
ここで目的の生物のゲノムリファレンスを取得する。今回はメダカHdRr系統(ASM223467v1 (GCA_002234675.1))にする

DNA(FASTA)というリンクをクリック、
このリストのdna.toplevel.fa.gzというファイルをクリックしてダウンロード!(またはcurlコマンドを使用)
これをgunzipコマンドで解凍(クリックして解凍すると拡張子がばぐる)
*必ずls -lでダウンロードしたファイルサイズを確認してHP上のサイズと一致してるか確認!たまにServerがばぐってファイルの一部がDLできていない時がある
ついでにマッピングには必要ないがゲノム配列に対応したアノテーションデータ(GFF/GTF)もダウンロード、解凍しておく
curl -OL http://ftp.ensembl.org/pub/release-104/fasta/oryzias_latipes/dna/Oryzias_latipes.ASM223467v1.dna.toplevel.fa.gz
curl -OL http://ftp.ensembl.org/pub/release-104/gtf/oryzias_latipes/Oryzias_latipes.ASM223467v1.104.gtf.gz
gunzip Oryzias_latipes.ASM223467v1.dna.toplevel.fa.gz
gunzip Oryzias_latipes.ASM223467v1.104.gtf.gz
ここまでに
Oryzias_latipes.ASM223467v1.104.gtf
Oryzias_latipes.ASM223467v1.dna.toplevel.fa
cab_liver_1_1.fastq.gz
cab_liver_1_2.fastq.gz
cab_liver_5_1.fastq.gz
cab_liver_5_2.fastq.gz
というファイルができた、これらをDesktop直下のTEST_RUNというディレクトリに収納
2.4 トリミング&クオリティチェック
ここでは生データの下処理を行う.低クオリティ配列の除去とアダプター配列の除去
生データをデスクトップ直下のTEST_RUNというフォルダに置いている
TEST_RUNへ移動(cd ~/Desktop/TEST_RUN)する
trim_galore --q 20 --illumina --fastqc --trim1 --paired cab_liver_1_1.fastq.gz cab_liver_1_2.fastq.gz
trim_galore --q 20 --illumina --fastqc --trim1 --paired cab_liver_5_1.fastq.gz cab_liver_5_2.fastq.gz
を実行
*結構時間がかかる
--q 20 クオリティの低いリードを除去、デフォルトの値20を採用
--illumina illuminaのアダプター配列13bpを除去
--trim1 3'末端の1bpを除去、クオリティが低いので
--fastqc内蔵のfastqcを動かしてQCを行う、QC結果はhtml形式で出力される
--pairedペアエンド方式でシーケンスしたことを指定
正常に作動したっぽいので生成ファイルができていた。

htmlファイルを開くとクオリティチェックの結果が見れる
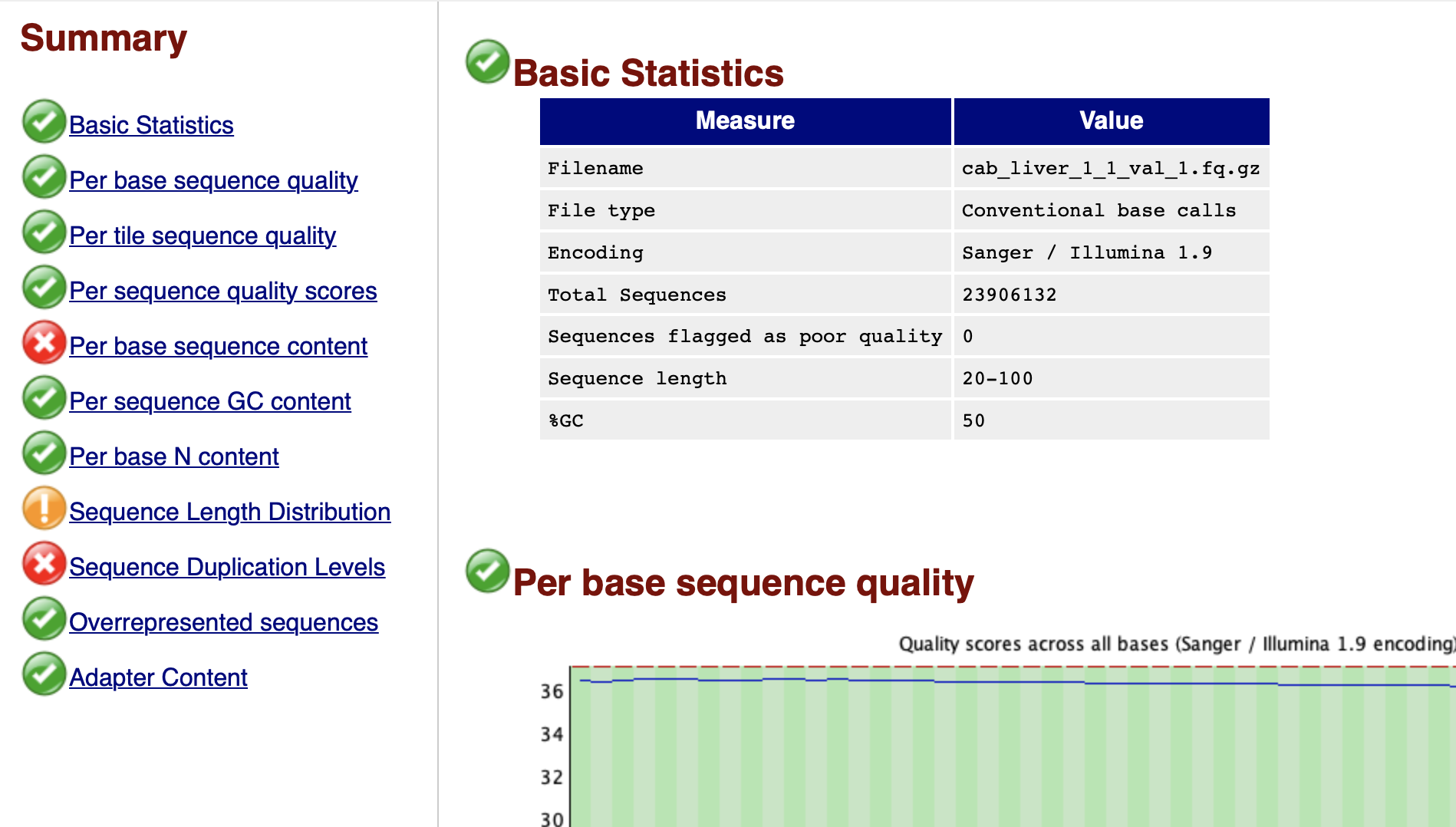
Summaryに項目が並ぶ。
per base sequenceはRNA-Seqでは必ず❌になるらしい
Sequence Length Distribution とSequence Duplication Levelsについては他のRNA-Seqの公開クオリティスコアでも❌になってたりしたので今回は知らんぷり(後で調べる)
生成された_val_1.fq.gzと_val_2.fq.gzファイルがトリミング済みの配列となる
2.5 マッピング
2.5.1 リファレンスインデックスの作成
Oryzias_latipes.ASM223467v1.dna.toplevel.faはそのままマッピングに使うことはできない
そこでインデックスファイルを作成する
インデックスファイルはマッピングツールごとに様式が異なるので都度作成する(hisat2、bowtie2用にそれぞれ作成するみたいに)
TEST_RUNへ移動(cd ~/Desktop/TEST_RUN)する
インデックスファイルをビルド,一度インデックスファイルを作っておけば次からメダカでマッピングする時は使いまわせる(hisat2にインデックス名が保存されているかは不明,今度時間をあけてマッピングをやってみる)
ビルドはhisat2-build input_genome.fa index_nameで実行
今回は
hisat2-build Oryzias_latipes.ASM223467v1.dna.toplevel.fa ASMol_index
を実行(約11分)
8つのインデックスファイルが生成される,ASMol_indexはこれらをまとめたオブジェクト(?,多分)
これでマッピング先のリファレンスは準備完了
2.5.2 マッピング
下処理したRNA-seqデータをインデックス化したリファレンスゲノムにマッピングする
hisat2 -p 8 -x ASMol_index -1 cab_liver_1_1_val_1.fq.gz -2 cab_liver_1_2_val_2.fq.gz -S cab1_out.sam
hisat2 -p 8 -x ASMol_index -1 cab_liver_5_1_val_1.fq.gz -2 cab_liver_5_2_val_2.fq.gz -S cab5_out.sam
-p 8でthreads数指定(このマシンは8コアなので8)
-x index_nameでindex_nameを指定
-1 left_reads.fq.gz -2 right_reads.fq.gzでペアエンドのリードを指定、.fa形式でも指定できるはず*もちろんtrim済みのやつ
-S out.samでsam形式で出力するファイル名を指定
結果
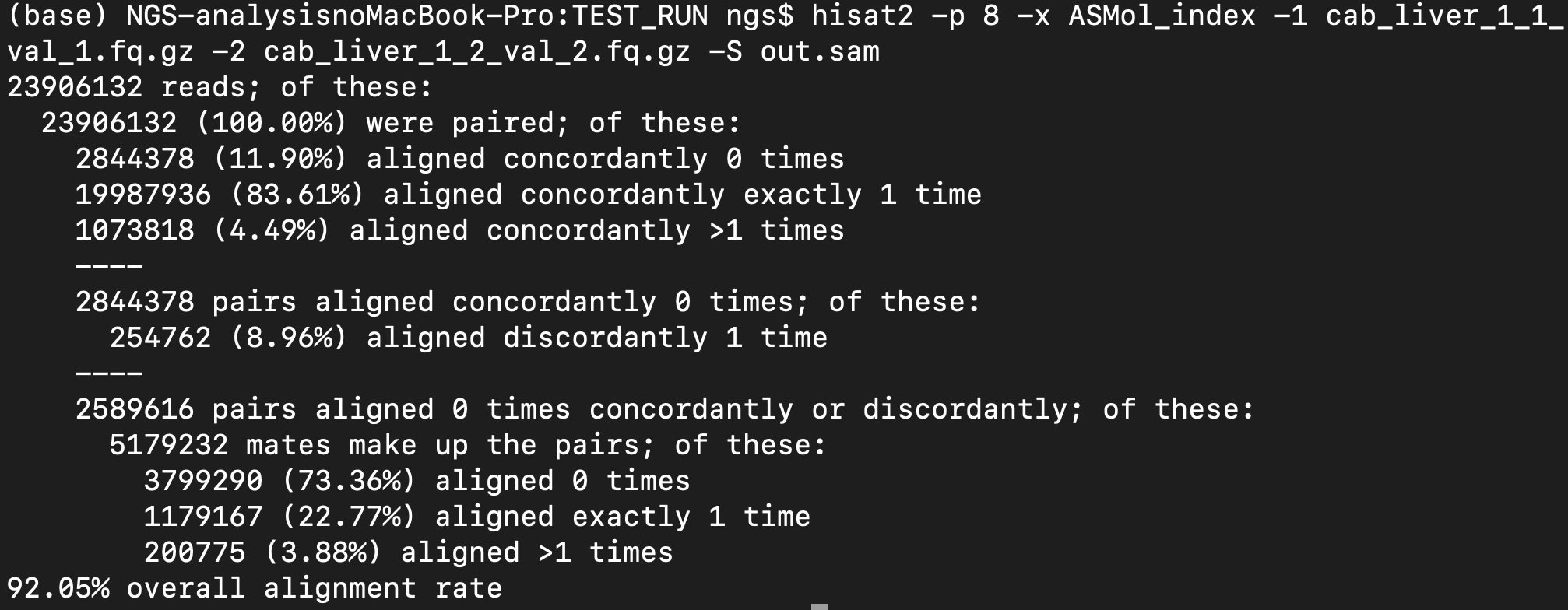
83.61%のリードが1回マッピングされた
4.49%のリードが2回以上マッピングされた
→89.10%のマッピング率、まあこんなもんか、ちょっと低いか?
*マッピング率が低いと他の生物種ゲノムのコンタミが疑われる、例えば微生物など。野外サンプルや表皮・腸管を含むサンプルでは注意すること
2.6 発現定量準備(ソート済BAMファイルの作成)
先ほどのマッピングの結果は.samファイルに収納されている(今回はout.samとして出力した)
StringTieで発現定量をするためにSAMtoolsを使ってマッピング結果をソートしてBAMファイルに変換する
samtools sort -@ 8 -O bam -o cab1_out.sort.bam cab1_out.sam
samtools sort -@ 8 -O bam -o cab5_out.sort.bam cab5_out.sam
を実行
-@ でメモリを指定、オプションなのでなくてもいい
-O bambamファイルで出力することを指定
-o output_file_nameで出力ファイル名を指定
(追記21.07.07)
同様の環境を整えた別マシンでsamtoolsを動かしたところ依存関係がばぐって動かなかった(bioconductor関連のパッケージを入れたのでそれかもしれない)
→conda install samtoolsで再インストールしたら正常に動いた
2.7 StringTieを使った発現定量
2.3でダウンロード&解凍したアノテーションGTFファイル(今回はOryzias_latipes.ASM223467v1.104.gtf)とマッピング結果(今回はソート済BAMファイル)を使って各遺伝子の発現定量をStringTie行う。今回はstringtieのオプションである-e -B メソッドを用いてballgown用(DEG解析ツール)の出力ファイルを作る。ただし今回はstringtieの後にballgownは使わない、なぜならballgownは試験区内のサンプルの分散を計算しないと進めないようで、今回はn=1のサンプルなのでballgownを使えない。そこでballgown用の出力ファイルをcsvファイルに変換して(prepDE.py)edgeRを用いてDEG解析を行うことにする。複製ありのballgownを使ったDEG解析はまた今度....
ではstringtieで発現量を取得する。
stringtie Cab_liver1_out.sort.bam -e -B -p 4 -o Cab_liver1.gtf -G Oryzias_latipes.ASM223467v1.104.gtf
stringtie Cab_liver5_out.sort.bam -e -B -p 4 -o Cab_liver5.gtf -G Oryzias_latipes.ASM223467v1.104.gtf
-o outfile_name 出力ファイル名の指定、gtf形式にすること
-G annotation_GTF_filename 参照するアノテーション情報リファレンス
sampleごとに出力ファイルをまとめておこう
tdata.ctabファイルを開いてみると以下のように、
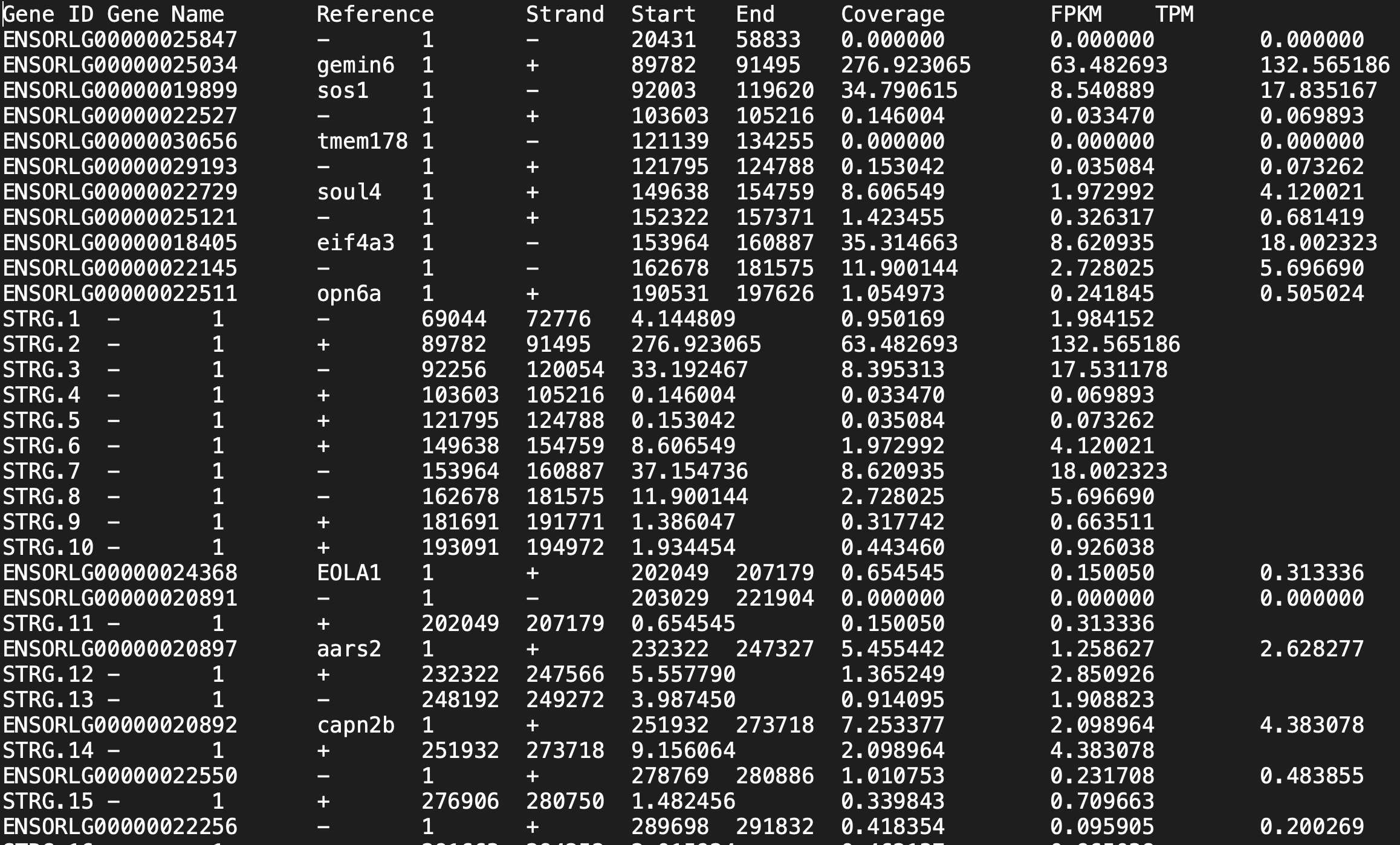
これでメダカのRNA-Seq生データから各遺伝子の発現定量まで完了した!
このファイルをedgeRに入力するためにcsvファイルへと変換する
変換にはprepDE.pyというスクリプトを実行する。少しややこしい
まずはprepDE.pyをインストール
curl -O https://ccb.jhu.edu/software/stringtie/dl/prepDE.py
次にこのスクリプトはpython2系で書かれているのでpython3で動くように変換し、実行する。この時prepDE.pyがおかれたディレクトリにcab1とcab5というフォルダを置き、その中にstringtie出力ファイルをまとめておく。
2to3 -w prepDE.py
python3 prepDE.py
prepDE.pyが正常に走るとgene_count_matrix.csvとtranscript_count_matrix.csvという2つのcsvファイルが生成される。
以降の解析ではこのcsvファイルとRStudioを使ってDEG解析を行う。遺伝子の発現を知りたい場合はgene_count_matrixを、転写産物単位の発現量を知りたい場合はtranscript_count_matrixを使う。
Rstudioを開いて、ワーキングディレクトリ(ballgownやhisat2の出力ファイルでごちゃごちゃしていると思うので整理しておく)へ移動する。
今回はanalysisフォルダのballgownというフォルダに移動する。ballgownには先ほど生成したgene_count_matrix.csvが置いてある。
ワーキングディレクトリの移動はsetwd(ディレクトリのパス)で行う。
edgeRで正規化とDEG検出を行う。今回はbiological replicateがn=1(複製なし)なので以下のスクリプトを参照した。*biological replicateはn=3が推奨されており、そのようなデザインの場合はDEG検出にballgownツールも使用できる(逆に複製なしの場合はDEG検出のツールがかなり制限されている気がする)。
#参照は解析 | 発現変動 | 2群間 | 対応なし | 複製なし | edgeR(robinson, 2010);https://www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html#about_analysis_deg_2_unpaired_nasi
in_f <- "gene_count_matrix.csv" #入力ファイル名を指定してin_fに格納
param_G1 <- 1 #G1群のサンプル数を指定
param_G2 <- 1 #G2群のサンプル数を指定
param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定
#必要なパッケージをロード
library(edgeR) #パッケージの読み込み
#入力ファイルの読み込みとラベル情報の作成
data <- read.table(in_f, header=TRUE, row.names=1, sep=",", quote="")#in_fで指定したファイルの読み込み
data <- as.matrix(data) #データの型をmatrixにしている
data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成
#正規化とDEG検出
d <- DGEList(counts=data,group=data.cl)#DGEListオブジェクトを作成してdに格納
d <- calcNormFactors(d) #TMM正規化を実行
d <- estimateGLMCommonDisp(d, method="deviance", robust=TRUE, subset=NULL, verbose=TRUE) #モデル構築(common Dispersionを算出)
out <- exactTest(d) #exact test (正確確率検定)で発現変動遺伝子を計算した結果をoutに格納
#tmp <- topTags(out, n=nrow(data), sort.by="none")#検定結果を抽出
p.value <- out$table$PValue #p値をp.valueに格納
q.value <- p.adjust(p.value, method="BH")#q値をq.valueに格納
ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納
sum(q.value < param_FDR) #FDR閾値(q.value < param_FDR)を満たす遺伝子数を表示
#ファイルに保存(テキストファイル)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)#入力データの右側にp.value、q.value、rankingを結合した結果をtmpに格納
tmp<-data.frame(tmp) #dataframe型にしておく
tmp$ranking<-as.numeric(tmp$ranking) #numeric形にしておく
tmp<-tmp[order(tmp$ranking),]
write.table(tmp,"comparative_result.csv", sep=",", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
tmp$q.value<-as.numeric(tmp$q.value) #q.vqlueも実数型に変換しておく
DEG_tmp<- tmp[tmp$q.value < param_FDR,] #q.valueが0.05以下のものをDEGとして格納
write.table(DEG_tmp, "DEG_list.csv", sep=",", append=F, quote=F, row.names=F) #DEGだけを別ファイルで保存
#AUC値を計算(高いほどよい方法; ただの検証用) このへんはよくわからん
library(ROC) #パッケージの読み込み
param_DEG <- 1:sum(q.value < param_FDR) #DEGの位置を指定
obj <- rep(0, nrow(data)) #初期値として全てが0の(non-DEGに相当)ベクトルobjを作成
obj[param_DEG] <- 1 #DEGの位置に1を代入
AUC(rocdemo.sca(truth=obj, data=-ranking))#AUC計算
#次にDEG(q.valueがFDR=0.05以下の転写産物)を識別して MAplotを描いてみる (参照:https://kazumaxneo.hatenablog.com/entry/2017/07/11/114021)
table<- as.data.frame(tmp) #データフレーム型に変換
is.DEG <- as.logical(table$q.value<param_FDR) #tableからq.valueの行を取り出して0.05より低いものをis.DEGに収納
DEG.names<-rownames(table)[is.DEG] #is.DEGの名前を取得
png("MA-plot.png") #png形式で保存するためにデバイスを開く
plotSmear(out, de.tags=DEG.names, cex=0.3) #DEGとそれ以外を識別してMAplotを出力
dev.off() #デバイスを保存
write.table(DEG.names, "DEG_names.txt", sep=",", append=F, quote=F, row.names=F) #DEGの名前をテキストファイルで保存
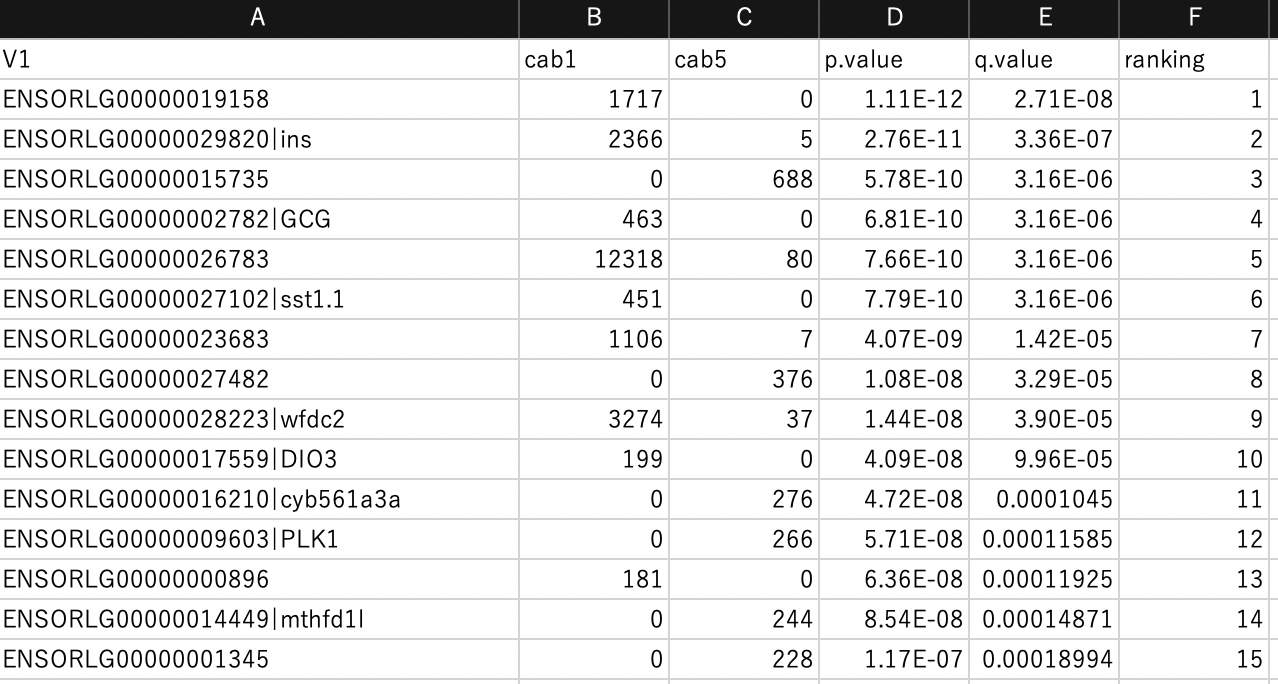
以上を実行するとsampleごとのリードカウント、p値、q値などがまとまったcsvファイル、そこからDEGだけが抽出されたcsvファイル、DEGの名前だけが抽出されたテキストファイル、MA-plot図が出力される。
DEGのリストとリード、p値、q値、q値のランキング(昇順)。正規化後のカウントも出して欲しい....
MA-plot。DEGは赤でプロットしている上記のコードのplotSmearのde.tags=DEG.namesで指定している
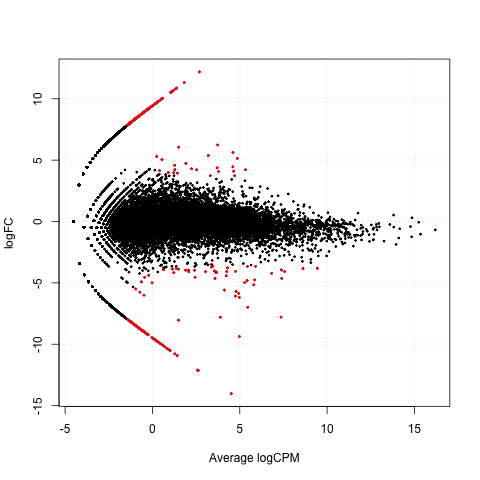
またサンプル間の発現変動遺伝子のリストを得ることができた(DEG_names.txt)。
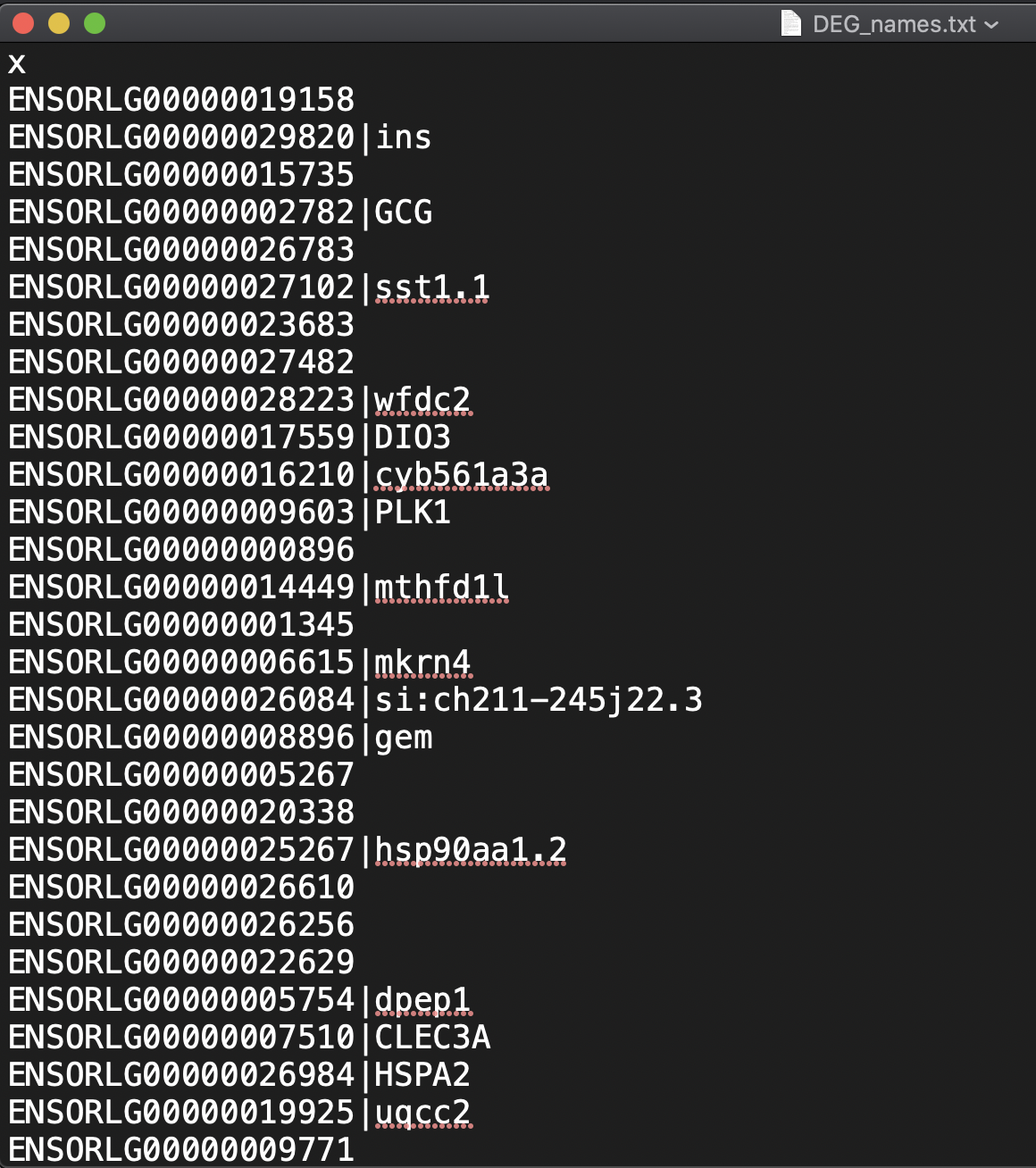
ただしこのテキストファイルを確認したところ、GENE-IDと遺伝子のsymbolが連結されているものとされていないものが混在していた。これでは後に行うDAVIDに入力ファイルとして認識されないので、GENE-ID(Ensemble GENE-ID)だけにする必要がある。
以下のスクリプトをGET_GENE-ID.pyとして保存してpython3で実行する。
with open('DEG_name.txt') as reader, open('DEG_GENE-ID.txt', 'w') as writer:
for line in reader:
line=line[:line.find("|")]
writer.write(line)
writer.write("\n")
実行するpython3 GET_GENE-ID.pyとDEG_names.txtからDEG_GENE-ID.txtが生成され、DEGのGENE-IDのみのテキストファイルが生成する。
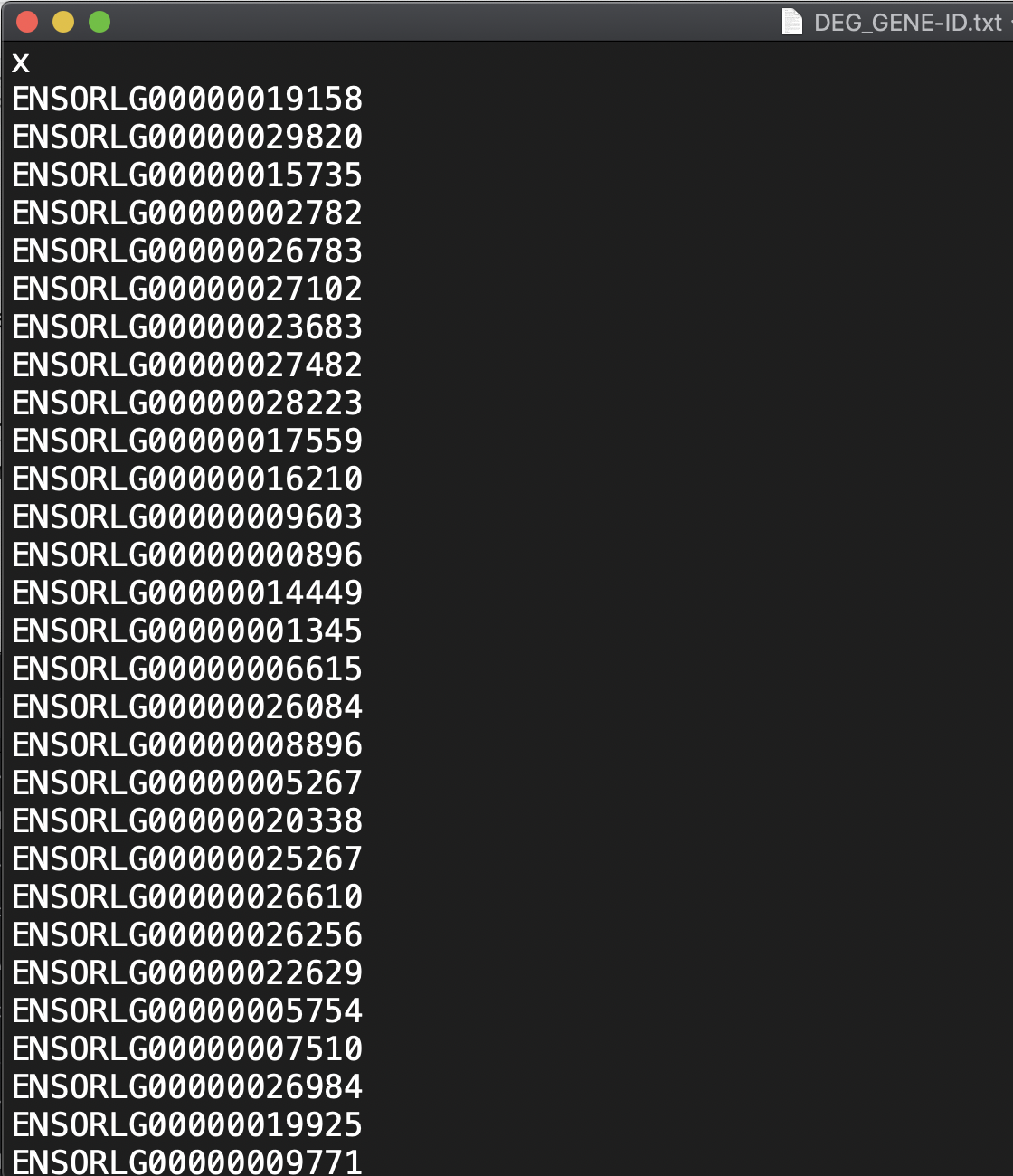
1行目にxって書いてあるけど気にしない(多分scvからtxtへ変換するときにヘッダーとして出てきてる気がする)
このファイルをDAVIDにアップロードして、GO解析やpathway解析などのエンリッチメント解析を行う。
DAVID:https://david.ncifcrf.gov

まず上のStart analysisをクリック,
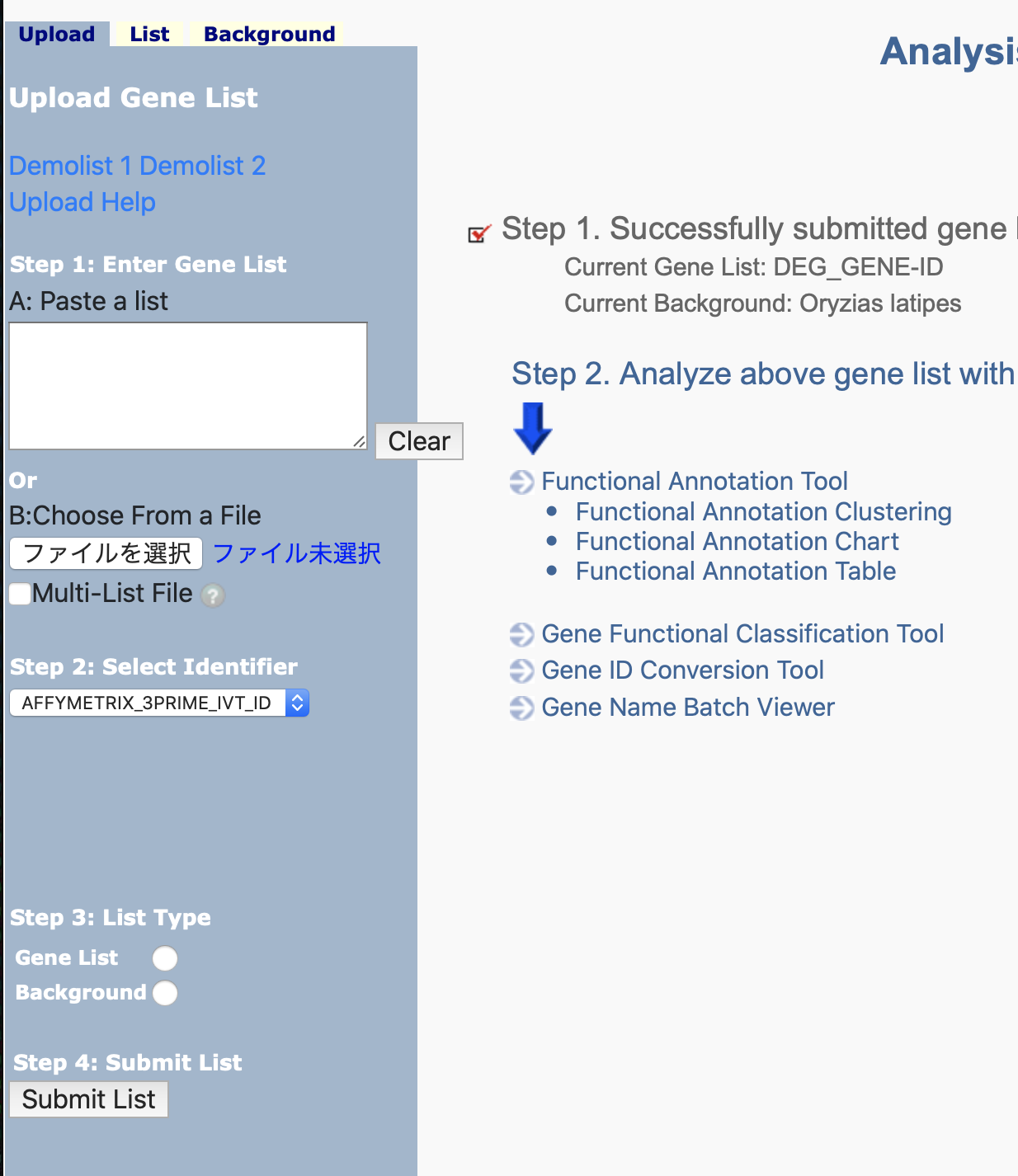
左のダイアログに先ほど生成したDEGのgene-IDのリストをアップロードし、step2:select identiferのところは自分のgene-IDがどこ由来かをチェックする。今回はensembleのサイトからダウンロードしたリファレンスやアノテーションファイルにマッピングして発現定量しているので、ensembleのgene-IDになっているはず。
step3:list typeはgene listで多分よいのでsubmit listをクリック。この後speceiesを選択してもう一度step1~3を入力すると各種解析の画面へ進むことができる。functional anotation toolをクリック。
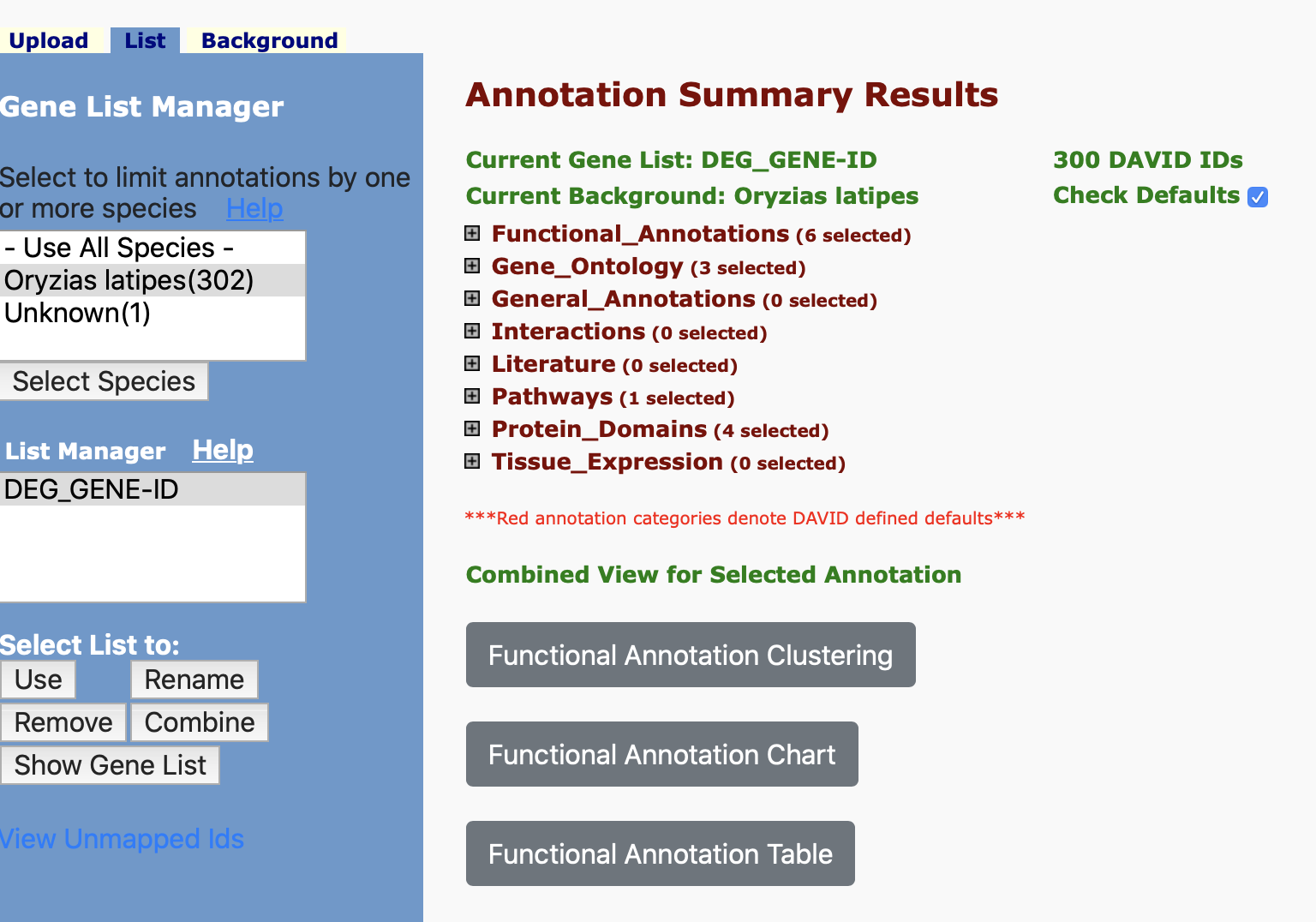
こんな感じに。gene onthology解析やpathway解析などが行える。
試しにPathwaysのKEGG-pathwayのchartを押すとfunctional annotation chartが、chart横の青いバーを押すとfunctional annotation tableが開かれる。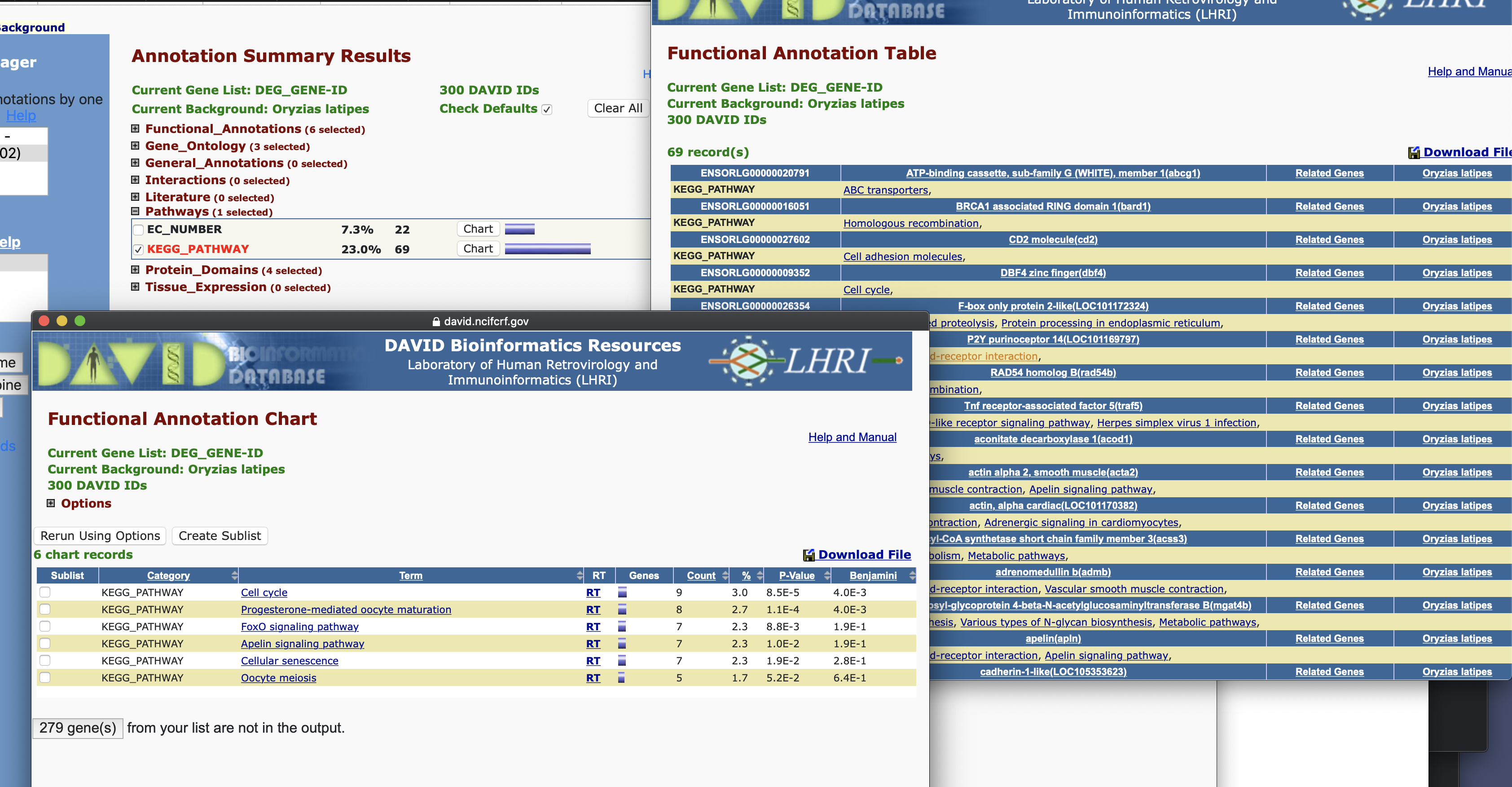
これらにはアップロードしたDEGがどのパスウェイに関与するかを示している。
DAVIDのさらなる使い方については、http://array.cell-innovator.com/?p=68 やhttps://togotv.dbcls.jp/20130528.htmlに詳しい。
試しにwntのパスウェイを図示してみた。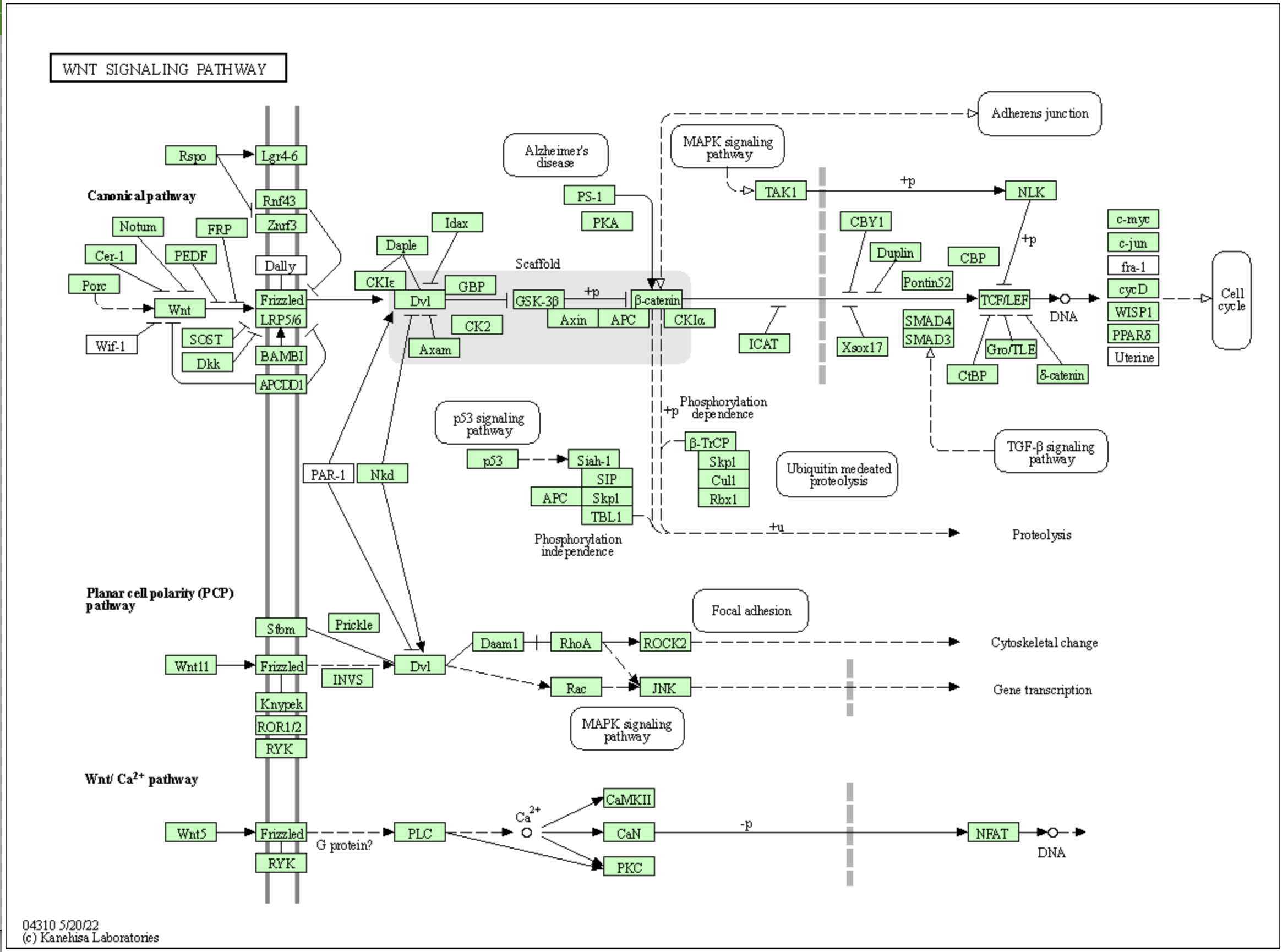
今回はRNA-Seqの一連の流れを実際にターミナルやウェブツールを用いて生データからエンリッチメント解析まで行った。