はじめに
こんにちは!バイオインフォマティクス系オタク修士学生の roadricefield です!
ChIP-seq や ATAC-seq などから得られたピーク領域を転写因子結合モチーフエンリッチメント解析にかけると本当に色々な転写因子がヒットしますよね. でも, 僕のような純粋生物学屋からすると「いや, 結局そのモチーフはどこに何個ぐらいあるんや!!」と興味のある転写因子の結合モチーフの実際の場所や個数を知りたくなってしまいます. 願わくばそのモチーフの周りのクロマチンの開閉状況なども合わせて確認して "Seeing is believing." などと言いながらしたり顔でゲノムブラウザ等で観察したいものです.
この記事では様々なモチーフに関連した解析用のツールを提供してくれている The MEME Suite の中の FIMO というソフトを使って上記の希望を叶えていきます.
FIMO は探したいモチーフの情報と DNA 配列を入力するとモチーフをスキャンしてくれるソフトです. それでは早速やってみましょう!
※ 「共同研究先から送られてきたピークの位置が書かれたファイル (BED ファイル) を持っていてこれに特定の転写因子のモチーフがどれくらいあるかだけ調べたいんだけどコマンドラインやったことないよぅ......(泣)」という方も大丈夫です!なんと, MEME はブラウザからボタンをクリックするだけでも使用できます! ブラウザでの使用方法だけを知りたい方は一気に「4. ブラウザでの FIMO の使用」まで飛んでください.
1. MEME のインストール
MEME は bioconda からインストールできます. 去年のアドベントカレンダーに私が投稿したこちらの記事の方法で環境構築をするなどして
conda install -c bioconda meme
で OK です. ためしに FIMO を起動してみましょう. ターミナルで fimo コマンドを実行してください.
roadricefield@ubuntu:~$ fimo
Usage: fimo [options] <motif file> <sequence file>
Options:
--alpha <double> (default 1.0)
--bgfile <background> (default from NR sequence database)
--max-stored-scores <int> (default=100000)
--max-strand
--motif <id> (default=all)
--motif-pseudo <float> (default=0.1)
--no-qvalue
--norc
--o <output dir> (default=fimo_out)
--oc <output dir> (default=fimo_out)
--parse-genomic-coord
--psp <PSP filename> (default none)
--prior-dist <PSP distribution filename> (default none)
--qv-thresh
--skip-matched-sequence
--text
--thresh <float> (default = 1e-4)
--verbosity [1|2|3|4] (default 2)
--version (print the version and exit)
When scanning with a single motif use '-' for <sequence file> to
read the database from standard input.
Use '--bgfile motif-file' to read the background from the motif file.
このようなマニュアルが表示されたら正常にインストールされています.
2. この記事で使用するデータのダウンロードと処理
2-1. どんなデータ?
この記事ではそれっぽいことをするために ENCODE project で取得されたこちらのマウスの制御性 T 細胞(アニメ, はたらく細胞で早見沙織さんが声をされているあの細胞です.)の ATAC-seq のデータをダウンロードしてこれを解析してみます. ATAC-seq はクロマチンの開閉状況をゲノム全体で調べるシークエンスです.
制御性 T 細胞については Sakaguchi, 2008, Cell などで紹介されていますがざっくり言うと免疫寛容を制御する細胞です. Foxp3 という転写因子が制御性 T 細胞の master regulator としてあまりにも有名 (Hori et al., 2003, Science) なので今回は制御性 T 細胞の ATAC-seq のピークの中に Foxp3 の結合モチーフを探してみましょう!
※ これから作成して FIMO で使用する ATAC-seq のピーク位置の DNA 配列 (Treg_ATAC_peaks.fa) をこちらにおいておきます. これを使えばわざわざマッピングやピークコールをする必要はありません.
2-2. データの落とし方
上記のこちらのページの以下の画像の "File details" を押してください.
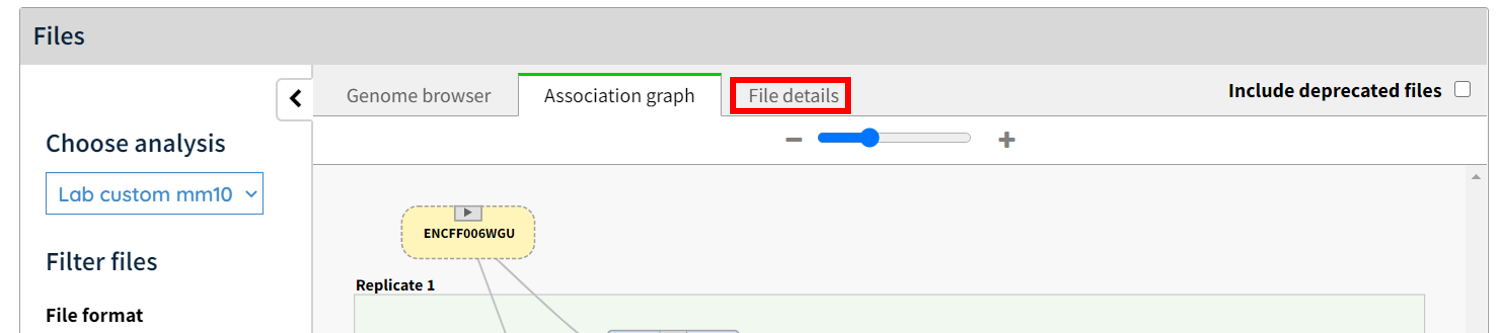
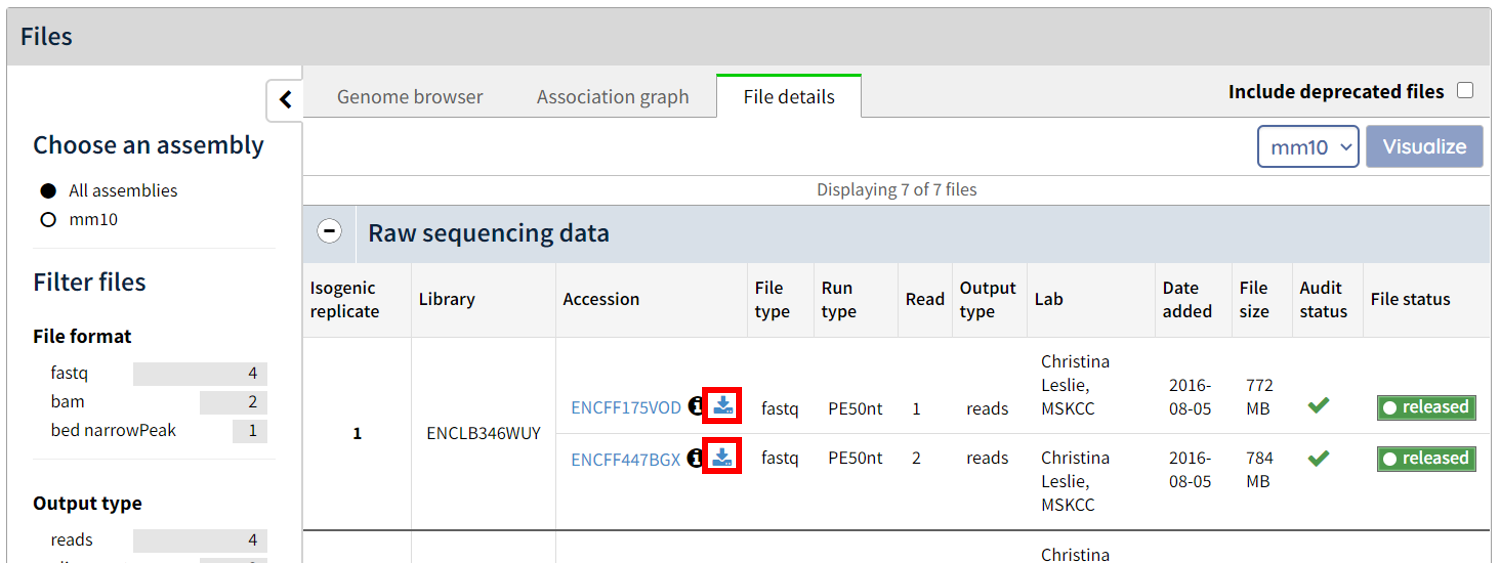
画面が切り替わって以下の画像の矢印2つを押して read1 (ENCFF175VOD.fastq.gz) と read2 (ENCFF447BGX.fastq.gz) をブラウザ経由でダウンロードします. 本当はこの下に replicate がもう一つ用意されていますが今回はこの replicate のみ解析します.
2-3. ダウンロードしたデータの処理
このデータは ATAC-seq のデータですが, 先程の私の記事と同じ方法で処理できます. ChIP-seq と ATAC-seq はデータの形式がほぼ同一です. こちらにアダプタートリミングからマッピングまでを行い, ピークコールと BigWig ファイルの作成までを一気に行うシェルスクリプトをおいておくので内部のパスを編集して使用してください. bowtie2 のインデックスは別途作成してください. こちらで説明しています. 今回の ATAC-seq のデータの処理はゼロからはじめるChIP-seq解析の内容と以下の点で異なります.
- シークエンスデータがペアエンドであること.
- ピークコールに HOMER の
findPeaksではなく MACS2 を使用すること.findPeaksでももちろんかまいませんが私が用意したスクリプトをご使用いただく場合は MACS2 となります. MACS2 も bioconda からインストールできますので$ conda install -c bioconda macs2でインストールしてください. - 用意したスクリプトでは ATAC-seq 特有の QC 方法を行っています. 詳しくはスクリプトのある Github ページの README をお読みください.
3. FIMO による Foxp3 結合モチーフのスキャン
得られた制御性 T 細胞の ATAC-seq のピーク, Treg_ATAC_peaks.bed の中に Foxp3 のモチーフがどこに何個, どれくらいのマッチングスコアで存在しているのかを FIMO で調べます.
3-1. ピーク位置の DNA 配列の取得
ピークの DNA 配列が必要なので bedtools の getfasta を使用します (コマンドの詳細はこちら).
bedtools も bioconda からインストールします.
conda install -c bioconda bedtools
それでは getfasta でピーク位置の DNA 配列の FASTA ファイルを引っこ抜きます.
bedtools getfasta -fi ${path to your mouse genome} -bed XXX/Treg_ATAC_peaks.bed -name | fold -w 50 > Treg_ATAC_peaks.fa
-fi の引数にはマウスのゲノムを一つの FASTA ファイルにまとめたものへのパスを, -bed の引数には Treg_ATAC_peaks.bed へのパスを指定してください. -name オプションをつけることでゲノム上での位置(chr, start, end)を FATSA のヘッダーに表示します.
上記コマンドでは getfasta の出力をパイプで受けて fold コマンドで 50 文字ずつ改行しています.
と, 言うわけで以下のようなファイルができたでしょうか?
>::chr1:4768521-4768616
AGCCTAAGAAATTTTAGGAAACAGTGTCACCTAGTGGTCACACACGGCAC
TGCAGAAATGTTAACTTGATTTCGTGTCCCATTTCACTTACAAag
>::chr1:4785460-4786037
TGCTTAGCATCCTTAACACTGGGCGCACGCGGAAGGGAGCCAGGCGCCCG
CACGCGTCCATCGCCAGAGTGACGCGGCCCCTGCACTCCCCGCACCGCCG
CCAAGCGTTTACCCGTTTTCTGGAGTTAGGACTGGGCTTCAGGTTGGCCA
3-2. JASPAR2020 から Foxp3 の結合モチーフをダウンロードする
今回は JASPAR2020 から Foxp3 の結合モチーフのモチーフファイルをダウンロードします.
JASPAR2020 でモチーフを検索する方法はこちらのTogoTVで解説してくださっているので御覧ください(こちらの動画は JASPAR2018のものですが2020も同じです).
JASPAR2020 にはマウスの Foxp3 のモチーフは登録されていませんでしたがヒトのものはありました. -> ID: MA0850.1
マウスの Foxp3 の結合モチーフを調べている Marson et al., 2007, nature の Fig.2b のロゴとほとんど一致しているのでヒトの FOXP3 の結合モチーフのモチーフファイルを使うこととします.
モチーフファイルのダウンロードはかんたん!以下の画像の MEME というボタンを押すだけ!

MA0850.1.meme というファイルがダウンロードされます.
MEME version 4
ALPHABET= ACGT
strands: + -
Background letter frequencies
A 0.25 C 0.25 G 0.25 T 0.25
MOTIF MA0850.1 FOXP3
letter-probability matrix: alength= 4 w= 7 nsites= 211 E= 0
0.350711 0.000000 0.649289 0.000000
0.070671 0.183746 0.000000 0.745583
0.871901 0.000000 0.000000 0.128099
0.925439 0.000000 0.039474 0.035088
0.767273 0.054545 0.178182 0.000000
0.000000 0.748227 0.113475 0.138298
0.921397 0.000000 0.078603 0.000000
URL http://jaspar.genereg.net/matrix/MA0850.1
中身はこのような A,T,G,C それぞれの出現頻度を表す行列になっています.
3-3. FIMO の実行
いよいよ FIMO を実行して 制御性 T 細胞のオープンクロマチン領域内の Foxp3 結合モチーフを探します.
fimo --o fimo_Foxp3_in_Treg_ATAC XXX/MA0850.1.meme YYY/Treg_ATAC_peaks.fa
--o オプションには結果を吐き出すディレクトリの名前をしてしてください. これを指定しない場合はカレントディレクトリに fimo_out というディレクトリが作成されてそこの中に結果が保存されます. 必須の引数はモチーフファイルへのパスとそのモチーフを探す FASTA ファイルへのパスです. この順で入力してください.
さて, 作成された fimo_Foxp3_in_Treg_ATAC の中を確認するとファイルが 5 個作成されていると思います. まずは fimo.html を開いてみましょう.

このように結果がわかりやすくまとめて表示されると思います.
私の環境では 1555 個ヒットしました. fimo の詳細なオプションなどについてはこちらを御覧ください.
3-4. モチーフの位置の BED ファイルを作成する.
最後に, 発見した Foxp3 のモチーフの位置を BED ファイルにしてみましょう.
やり方は簡単で, 出力ファイルのうち fimo.txt の start, end の列が各 DNA 配列の 5' 末端側から数えたときの start, end となっていますのでそれを使います.(ストランドの +/- は FIMO では気にする必要はありません.)
こちらに R スクリプト (Make_Foxp3_loci_bed.R) をおいておきます. R の使い方は私が書いたこちらの記事などを参考にしてください.
3-5. ゲノムブラウザで確認する
ATAC-seq の結果の BigWig ファイルと上記で作成した Foxp3 のモチーフの位置をゲノムブラウザで確認してみましょう.
Satb1 付近

Irf4 付近
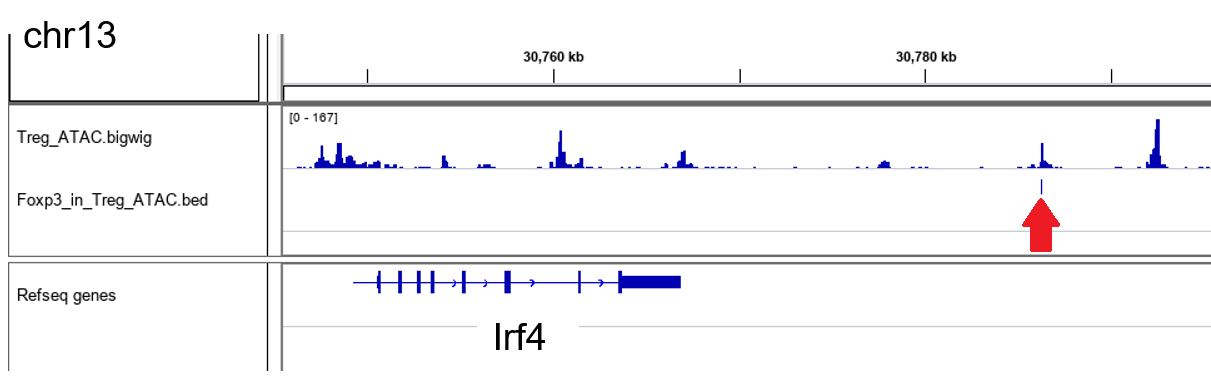
ATAC-seq のピークのちょうど中央付近に Foxp3 のモチーフがありますね!(思ったより少なかったなぁ~......)これらの遺伝子は Foxp3 の制御を受けている可能性があります.
3-6. モチーフ周りの各種シークエンスデータのシグナルを可視化する
おまけです. 発見したモチーフの位置における ChIP-seq のデータのシグナルなどを可視化したいということがあると思います. 今回はほとんど意味がありませんが発見した Foxp3 のモチーフ付近の ATAC-seq のシグナルを可視化してみます.
deeptools の computeMatrix と plotProfile を使用します.
computeMatrix reference-point -S XXX/Treg_ATAC.bigwig -R YYY/Foxp3_in_Treg_ATAC.bed -a 2000 -b 2000 -bs 1 --averageTypeBins sum --skipZeros -out Foxp3_ATAC_computeMatrix.gz
plotProfile -m Foxp3_ATAC_computeMatrix.gz --averageType mean --plotHeight 12 --plotWidth 12 --refPointLabel "Foxp3 motif" -z "" -out ATAC_at_Foxp3_motif.pdf --outFileNameData ATAC_at_Foxp3_motif.txt
こんな感じになります.
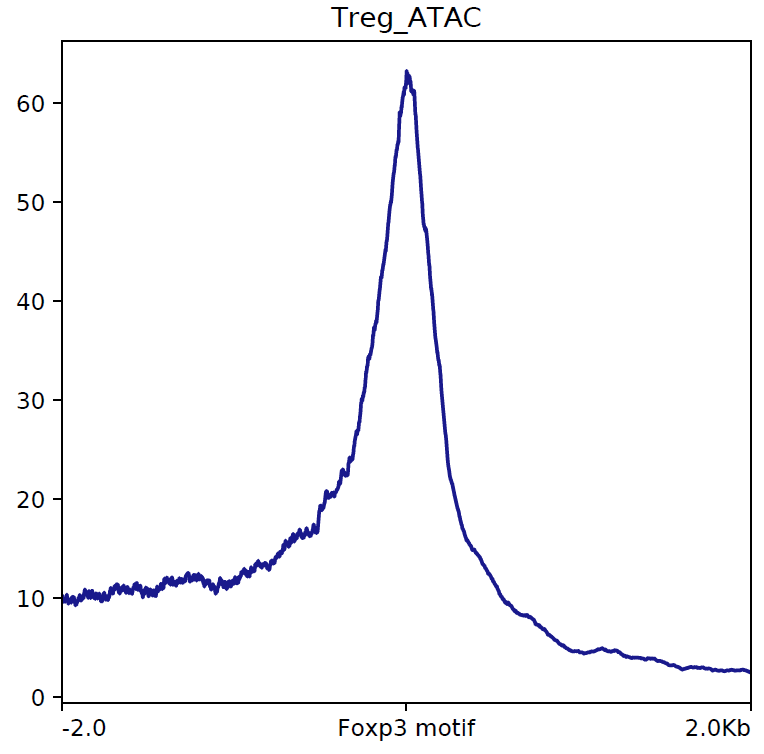
ATAC_at_Foxp3_motif.txt にもとの数値データが入っているのでこれを読み込んでもっときれいなグラフを作ることも可能です.
4. ブラウザでの FIMO の使用
FIMO はブラウザのみでも使用できます. ブラウザ版の FIMO はこちら

-
モチーフファイルをアップロードします.
-
配列をアップロードします. プルダウンメニューが最初は
Upload sequenceになっていませんので注意してください. -
Start Searchを押してください.
しばらくすると自動でページが遷移して結果をダウンロードするページに飛びます.
とってもかんたん!!しかしどうしても配列の FASTA ファイルが必要になります. ごめんなさい... BED ファイルの DNA 配列を引っこ抜く GUI のみで動くソフトをもしご存知の方がいらっしゃればコメントにてご教示いただけると幸いです.
The MEME Suite には FIMO の他にも便利なツールが多くあり, ブラウザ上でも動作します. みなさんも試してみてくださいね!
免責事項
本記事の正確性や妥当性につきまして,一切の保障はいたしません.また本記事を参考にして解析を行ったことで生じたあらゆる損害について,一切の責任を負いません.