この記事はバイオインフォマティクス Advent Calendar 2020の19日目の記事です
今年はCOVID19のパンデミックによりSARS-CoV-2の解析が注目されましたが、その解析はどのようなパイプラインになっているのでしょうか。Terraという解析プラットフォームにCOVID19のWorkSpaceがあったので、Terraを使ってみながら解析の流れを紹介したいと思います。
SARS-CoV-2ゲノムの構造
SARS-CoV-2ゲノムは30k塩基長の一本鎖RNAです。

すべてのβコロナウイルスゲノムに共通するオープンリーディングフレーム(ORF)と、スパイク(S)、エンベロープ(E)、マトリクス(M)、ヌクレオカプシド(N)タンパク質をコードしています。
Terra
TerraはGATKでおなじみのBroad InstituteとAlphabet傘下のVerilyが共同で開発したバイオメディカル研究のためのデータ解析プラットフォームです。
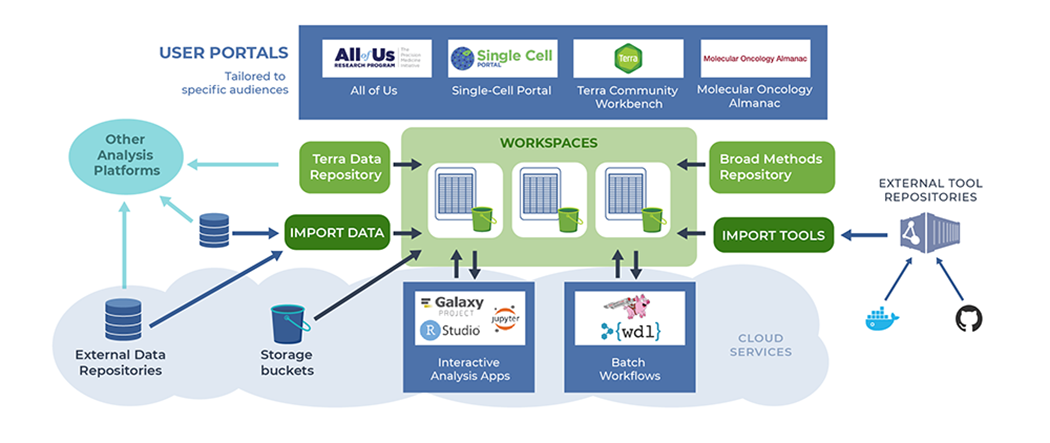
GCPのアカウントを用意すれば(従量課金ですが)、ワークフローを用いたデータ解析をGUIで実行することができ、データや結果の共有を簡単に行うことができます。
ワークフローはWDLで記述され、DockstoreとBroad Method Repositoryから追加することができます。
また、1000 GenomesやENCODE Project、その他様々なデータを参照することができるようになっています。
解析ワークフロー
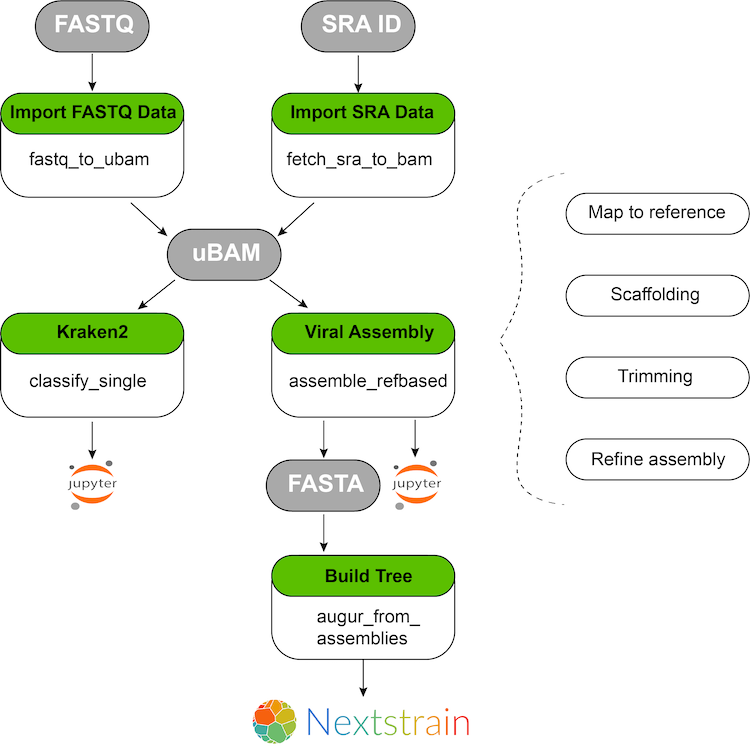
ウイルスゲノムを解析する場合にはDe novoアセンブリを用いたワークフローもありますが、ここではSARS-CoV-2のみを対象しているのでより効率的なリファレンス配列を用いた解析になっています。
-
SRAまたはFASTQファイルからuBAMファイルに変換する。
DATAタブであらかじめサンプルテーブルを作成しておく必要があります。SRAの場合はIDを、ファイルの場合はGoogle Storageへのファイルパスを指定します。 -
uBAMファイルからリファレンスゲノムを用いたアセンブルを行う。
1で作成されたuBAMファイルを指定して、リファレンスゲノムへのアライメントを行います。 -
系統樹を作成する。
AugurというツールでアセンブルされたFASTAから系統樹を作成します。
各ワークフローは以下のようなWDL形式で記述されていますが、Terra上では実行したいワークフローを選択するだけです。
version 1.0
import "../tasks/tasks_ncbi_tools.wdl" as ncbi_tools
workflow multi_Fetch_SRA_to_BAM {
input {
Array[String] SRR_accessions
}
scatter(SRA_ID in SRR_accessions) {
call ncbi_tools.Fetch_SRA_to_BAM {
input:
SRA_ID = SRA_ID
}
}
output {
Array[File] reads_ubam = Fetch_SRA_to_BAM.reads_ubam
Array[String] sequencing_center = Fetch_SRA_to_BAM.sequencing_center
Array[String] sequencing_platform = Fetch_SRA_to_BAM.sequencing_platform
Array[String] sequencing_platform_model = Fetch_SRA_to_BAM.sequencing_platform_model
Array[String] biosample_accession = Fetch_SRA_to_BAM.biosample_accession
Array[String] library_id = Fetch_SRA_to_BAM.library_id
Array[String] run_date = Fetch_SRA_to_BAM.run_date
Array[String] sample_collection_date = Fetch_SRA_to_BAM.sample_collection_date
Array[String] sample_collected_by = Fetch_SRA_to_BAM.sample_collected_by
Array[String] sample_strain = Fetch_SRA_to_BAM.sample_strain
Array[String] sample_geo_loc = Fetch_SRA_to_BAM.sample_geo_loc
Array[File] sra_metadata = Fetch_SRA_to_BAM.sra_metadata
}
}
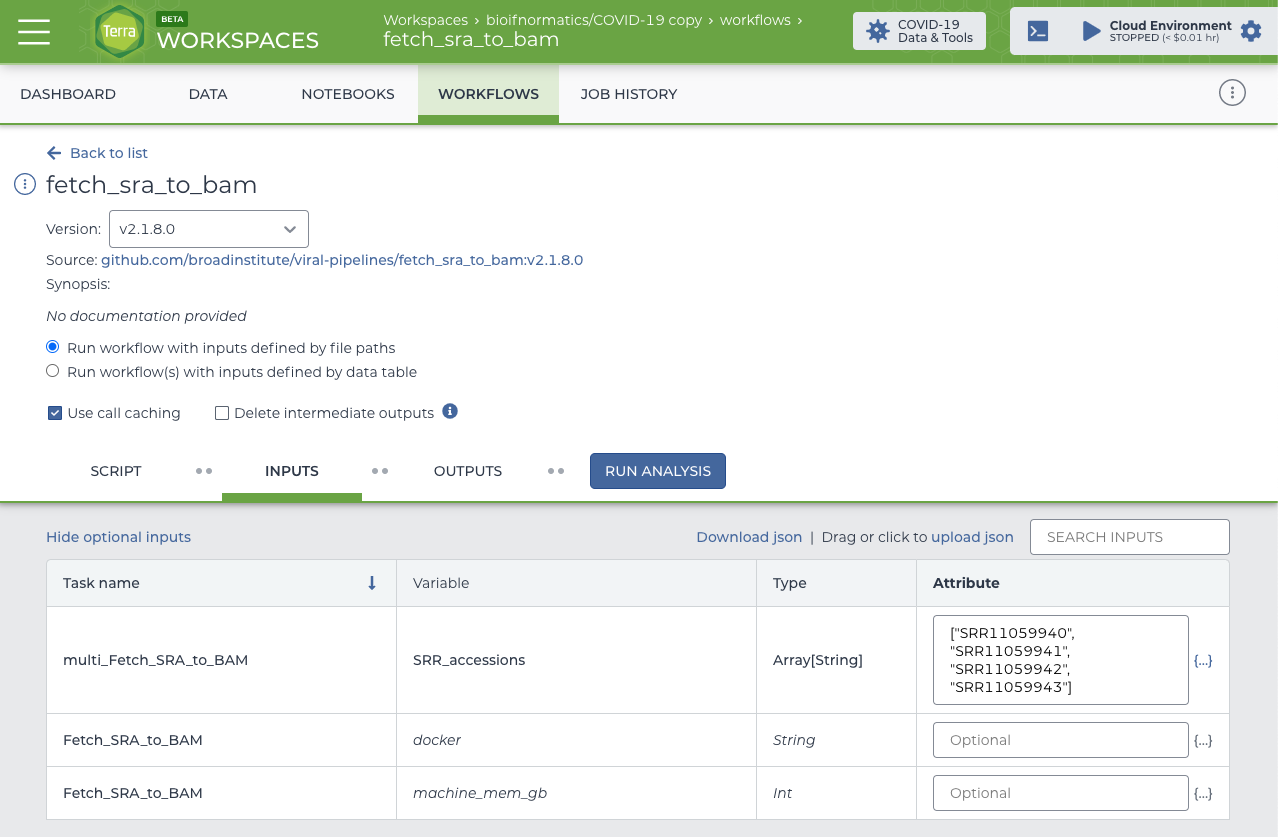
選択したWorkflowで入力ファイルを指定してRUN ANALYSISを押すと解析が実行されます。
fetch_sra_to_bamの例
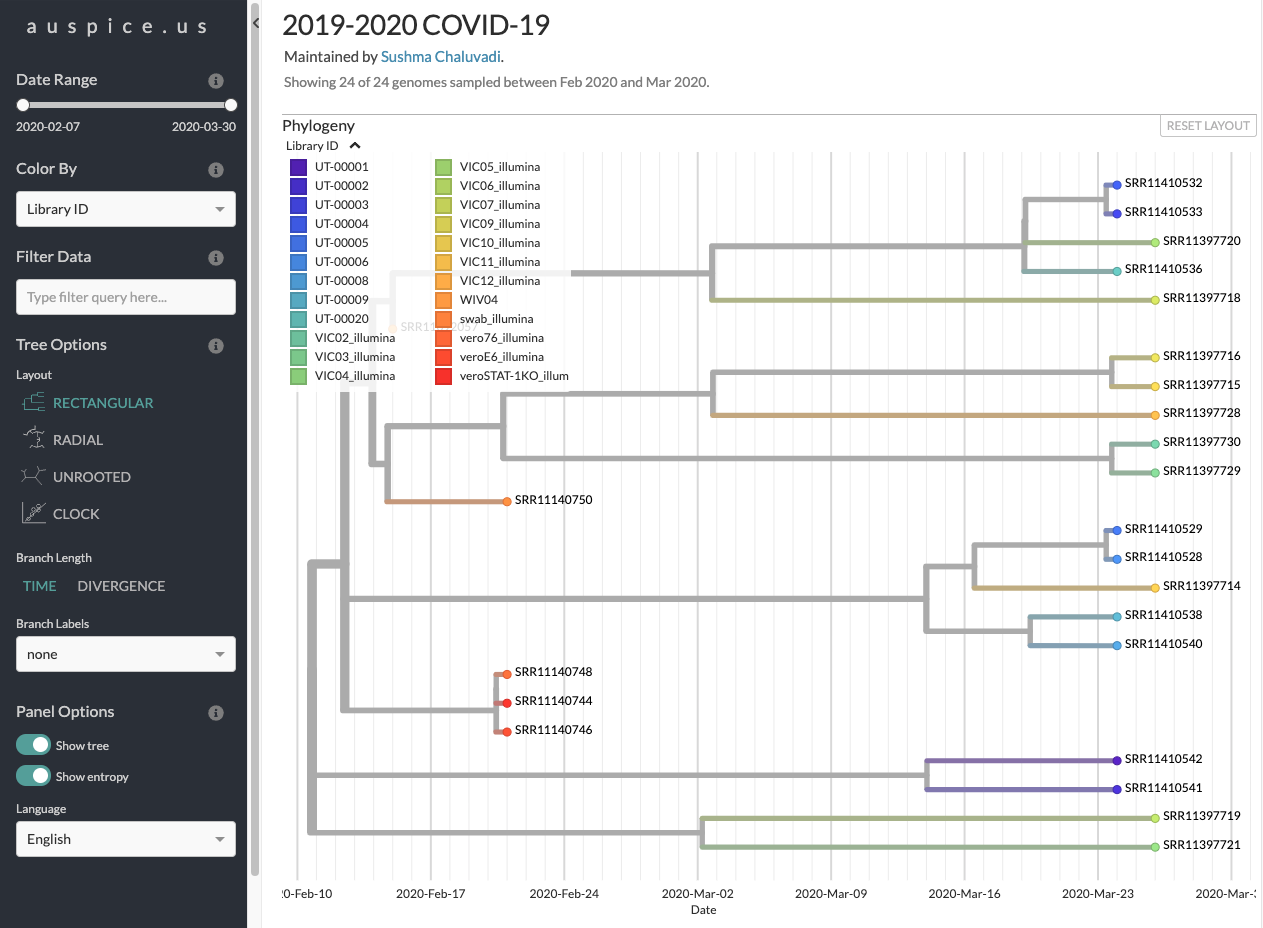
系統樹の描画
系統樹を描画するにはNextstrainを独自に立ち上げてもいいですが、auspice.usというサイトが公開されているのでそこに3で作成された系統樹のJSONファイルをDrag&Dropすることで以下のように描画することができます。
参考