はじめに
化合物探索において分子の部分構造(フラグメント)はとても重要です。例えば、以下のようなシーンで化合物のフラグメントは役に立ちそうですね。
- 分子の一部の構造を変えて物性が良くなる化合物を探したい
- 分子の一部の構造を変えていって似たような分子を作りたい(→ライブラリーを作りたい)
- ある分子とある分子の類似度をフラグメントを基準に評価したい(例:tanimoto係数)
この記事ではRDKitを用いた様々なフラグメント化の手法についてご紹介いたします。
Murcko
Murcko型分解:Murcko骨格(Murcko scaffold)に基づき、 分子のフラグメントへの分割と、骨格の取り出しを行なっています。Murcko scaffoldsでは、化合物から余分な側鎖を取り払い、"環構造"とそれらをつないでいる"Linker"のみで表現することでより単純な化合物表現を行います。
# ライブラリーのインポート
from rdkit import Chem
from rdkit.Chem import Draw
# RDKitのバージョン確認
from rdkit import rdBase
print('rdkit version: ', rdBase.rdkitVersion)
# ライブラリーのインポート
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit import rdBase
print('rdkit version: ', rdBase.rdkitVersion)
# 例となる分子を定義して検討していく
m1 = Chem.MolFromSmiles("NC1=NC2=C(C(OCC3OCCC3)=N1)N=CN2")
m1

# Murcko型分解により骨格を抜き出す
from rdkit.Chem.Scaffolds import MurckoScaffold
core = MurckoScaffold.GetScaffoldForMol(m1)
core
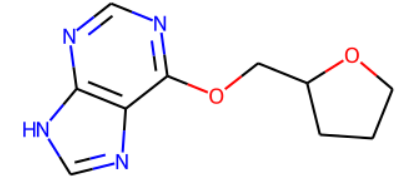
以下のコードで抜き出した骨格をさらに一般化することが可能です。
# 抜き出した骨格をさらに一般化する
fw = MurckoScaffold.MakeScaffoldGeneric(core)
fw
RECAP
RECAP(Retrosynthetic Combinatorial Analysis Procedure):生理活性物質に共通のフラグメント構造(11種類)に着目して、フラグメント化を行う
RECAPの元文献はこちらです。また11種類の構造は以下の通りです。

# 例となる分子を定義して検討していく
m2 = Chem.MolFromSmiles("CN(OC(C)=O)C1=CC=C(C2=CC=CC(O)=C2)C=C1")
m2

RECAPではもともとの分子を「root(根)」として考え、分解したフラグメントを「children」と言い、RECAPのルールに従ってこれ以上分解できないフラグメントを「leaf(葉)」と呼びます。まず元の分子を分解して第1段階のフラグメント化を行います。
from rdkit.Chem import Recap
# 化合物の分解
decomp = Recap.RecapDecompose(m2)
# 子ノードの情報の表示
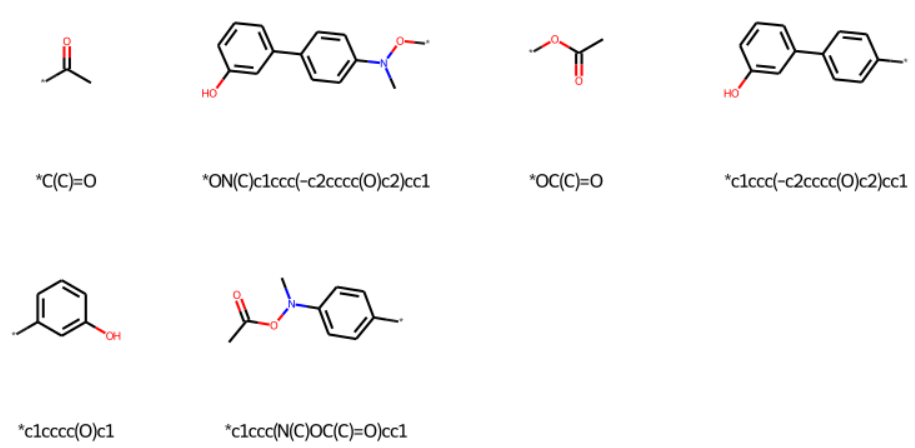
decomp.children
{'*C(C)=O': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfdcd0>,
'*ON(C)c1ccc(-c2cccc(O)c2)cc1': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfdca0>,
'*OC(C)=O': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfdbe0>,
'*c1ccc(-c2cccc(O)c2)cc1': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfdb80>,
'*c1cccc(O)c1': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfdac0>,
'*c1ccc(N(C)OC(C)=O)cc1': <rdkit.Chem.Recap.RecapHierarchyNode at 0x28be1bfde50>}
子のノードは辞書型として保持されます。
# 子ノードのフラグメントの描画
first_gen = [node.mol for node in decomp.children.values()]
Draw.MolsToGridImage(first_gen, molsPerRow=4, legends=[Chem.MolToSmiles(m) for m in first_gen])
RECAPによってどのように分解されたかを以下に示します。

11種類のフラグメントのうち、2(赤色),3(青色),10(緑色)のフラグメントに基づいてそれぞれ色分けされたフラグメントが生成していることが分かります。
子ノードの2つ目のフラグメントに着目してさらに分解していきます。
# 着目するフラグメントの描画
fg1 = first_gen[1]
fg1
# 化合物の分解
decomp_fg1 = Recap.RecapDecompose(fg1)
second_gen = [node.mol for node in decomp_fg1.children.values()]
Draw.MolsToGridImage(second_gen, molsPerRow=4, legends=[Chem.MolToSmiles(m) for m in second_gen])

これによって第2段階のフラグメント化が実行され、子ノードの子ノードが表示されたことになります。さらに2つ目のフラグメントを分解していきます
# 着目するフラグメントの描画
sg1 = second_gen[1]
sg1
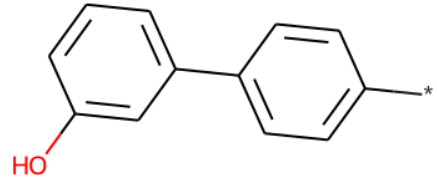
# 化合物の分解
decomp_sg1 = Recap.RecapDecompose(sg1)
third_gen = [node.mol for node in decomp_sg1.children.values()]
Draw.MolsToGridImage(third_gen, molsPerRow=4, legends=[Chem.MolToSmiles(m) for m in third_gen])
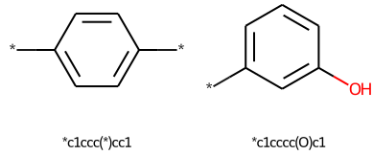
これらのフラグメントは分解することが出来ないので、「leaf(葉)」の扱いになります。試しに1つ目のフラグメントを分解すると空の辞書が返ってくることが分かります。
tg1 = third_gen[1]
decomp_tg1 = Recap.RecapDecompose(tg1)
decomp_tg1.children.values()
dict_values([])
leafを得る場合にはGetLeavesメソッドを使うのが便利です。
leaves = [leaf.mol for leaf in decomp.GetLeaves().values()]
Draw.MolsToImage(leaves, legends=[Chem.MolToSmiles(m) for m in leaves])

こちらを参考にSMILESでRECAPツリーを書いてみます。
# RECAPツリーを描く関数の定義
def get_leaves(recap_decomp, n=1):
for child in recap_decomp.children.values():
print('\t'*n+child.smiles)
if child.children: ##さらなるフラグメント化のチェック
get_leaves(child, n=n+1)
def get_recap_tree(mol):
recap = Chem.Recap.RecapDecompose(mol)
print(Chem.MolToSmiles(mol))
get_leaves(recap)
# m2に対してRECAPツリーを描く
get_recap_tree(m2)
CC(=O)ON(C)c1ccc(-c2cccc(O)c2)cc1
*C(C)=O
*ON(C)c1ccc(-c2cccc(O)c2)cc1
*O*
*c1ccc(-c2cccc(O)c2)cc1
*c1ccc(*)cc1
*c1cccc(O)c1
*c1cccc(O)c1
*ON(C)c1ccc(*)cc1
*O*
*c1ccc(*)cc1
*OC(C)=O
*O*
*C(C)=O
*c1ccc(-c2cccc(O)c2)cc1
*c1ccc(*)cc1
*c1cccc(O)c1
*c1cccc(O)c1
*c1ccc(N(C)OC(C)=O)cc1
*C(C)=O
*ON(C)c1ccc(*)cc1
*O*
*c1ccc(*)cc1
*OC(C)=O
*O*
*C(C)=O
*c1ccc(*)cc1
どのフラグメントも分解されて、最終的にleafの4つのいずれかに集約されることが分かります。
BRICS
BRICS(Breaking of Retrosynthetically Interesting Chemical Substructures):RECAPでは11種類のフラグメントを考えて切断しましたが,BRICSではさらに切断箇所を増やして16種類のフラグメントを考えます。
RECAPでフラグメント化した分子と同じ分子をここでもフラグメント化していきます。
# 例となる分子を定義(再掲)
m2 = Chem.MolFromSmiles("CN(OC(C)=O)C1=CC=C(C2=CC=CC(O)=C2)C=C1")
m2

# BRICSによるフラグメント化
from rdkit.Chem import BRICS
frag = BRICS.BRICSDecompose(m2)
frag
{'[1*]C(C)=O', '[16*]c1cccc(O)c1', '[3*]ON(C)c1ccc([16*])cc1'}
# フラグメントの表示
f_mols = BRICS.BRICSDecompose(m2, returnMols=True)
Draw.MolsToGridImage(f_mols, molsPerRow=3)
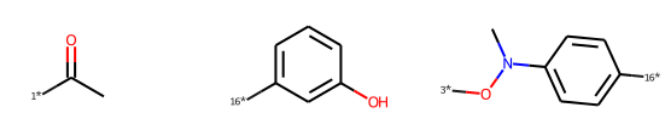
共通部分骨格(最大共通部分骨格MCS)
こちらの投稿にまとめていますので参考にしてください。
他のフラグメント化手法
環構造に含まれる/含まれない原子間の結合を切断し、環構造とその他に分解する。
# 例となる分子を定義して検討していく
m3 = Chem.MolFromSmiles("CC(C)C1=CC=CC(C(C2=CC=C(C(O)=CC(/C=C/C3=CC=NC3)=C4N)C4=C2)=O)=C1")
m3
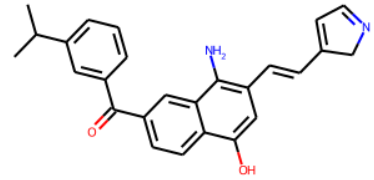
のちほど切断箇所の番号が必要になるので、分子内の原子に番号を付与します。
# 番号と原子を同時に指定するように関数定義
def mol_with_atom_index(mol):
atoms = mol.GetNumAtoms()
for idx in range(atoms):
mol.GetAtomWithIdx(idx).SetProp('molAtomMapNumber', str(mol.GetAtomWithIdx(idx).GetIdx()))
return mol
# m3に適用する
mol_with_atom_index(m3)
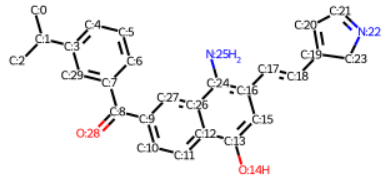
# 切断個所の明示
bis = m3.GetSubstructMatches(Chem.MolFromSmarts('[!R][R]'))
bis
((1, 3), (8, 7), (8, 9), (14, 13), (17, 16), (18, 19), (25, 24))
# 切断個所に該当する結合インデックスの抜き出し
bs = [m3.GetBondBetweenAtoms(x,y).GetIdx() for x,y in bis]
bs
[2, 7, 8, 13, 16, 18, 24]
# フラグメントの表示
fragments = Chem.FragmentOnBonds(m3,bs)
fragments
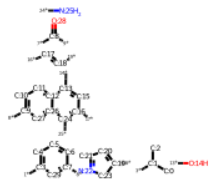
フラグメントが小さくなってしまったので描画しなおします。
from rdkit.Chem.Draw import IPythonConsole, MolsToGridImage
MolsToGridImage(Chem.GetMolFrags(fragments, asMols=True))
