はじめに
RDKitで分子の記述子を作っていく方法を紹介します。この方法を使って分子を数値化できれば、機械学習で予測モデルを構築することができます。イケメン Mas Kotさんがコッテコテの関西弁でRDKit記述子をええ感じに解説しとったので(こちらの投稿)ワイも負けじと関西弁で対抗していくで!イケメン Mas Kotさんは自ら関数を定義してのgithubにコードを載せてリンクするっちゅう高尚なことやってはるけど、この投稿ではRDKitのモジュールを使いこなしていくで。あっ、もちろんどっかのサイトのコピペで終了やなくて、3次元構造に紐づく記述子を計算するっちゅうオリジナリティも発揮してるさかい、最後まで見ていってくれよな。ほな、いってみよか!
RDKit記述子
記述子とは分子の性質や特性などを示すものやで。機械学習モデルは一般的にはy=f(x)という式で表されるけど、分子はそのままやったら予測モデルに代入できへんやろ?(x=ベンゼンを式に代入して・・・とかなかなか無理難題やろ?)だから記述子という数値に変換することで機械学習モデルでも分子を扱うことが出来るちゅうわけや。
Pythonでの実装
# ライブラリのインポート
import pandas as pd
from rdkit import Chem
from rdkit.Chem import Descriptors
from rdkit.Chem import AllChem
from rdkit.Chem import rdMolDescriptors
みんなに参考にしてもらおう思ったら、バージョンぐらいちゃんと書かんとな~
from rdkit import rdBase
print('rdkit version: ', rdBase.rdkitVersion)
rdkit version: 2020.09.1
次にデータを読み込んでいくんやけど、今回は以下のようなSMILESが書かれたcsvファイルを読み込むという想定でいくで。マテリアルズインフォマティクスの世界やったらよくあるパターンやろ。知らんけど。
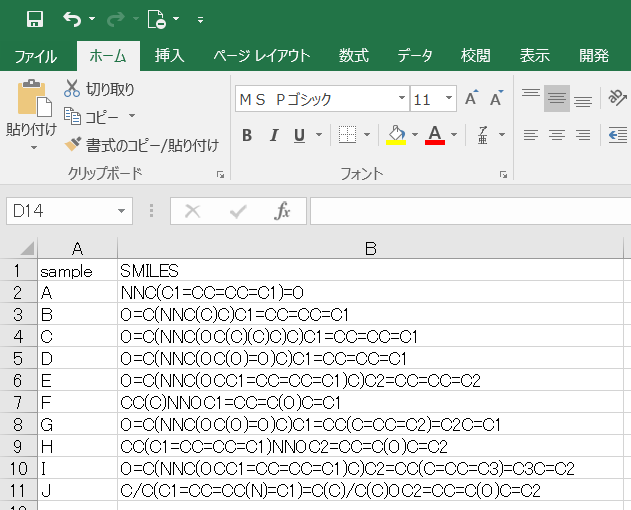
# ファイルの読み込み
data=pd.read_csv("sample_data.csv")
data.head()
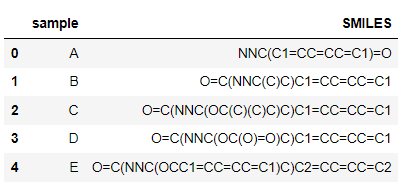
あとでこっそり使うので、SMILESをリストとして持っておくんやで!
# SMILESをリスト化しておく
smiles = list(data["SMILES"])
こちらのサイトを参考に(丸パクリ)して記述子を計算する関数を定義していくで。
# 記述子を計算する関数の定義
def getMolDescriptors(mol, missingVal=None):
''' calculate the full list of descriptors for a molecule
missingVal is used if the descriptor cannot be calculated
'''
res = {}
for nm,fn in Descriptors._descList:
# some of the descriptor functions can throw errors if they fail, catch those here:
try:
val = fn(mol)
except:
# print the error message:
import traceback
traceback.print_exc()
# and set the descriptor value to whatever missingVal is
val = missingVal
res[nm] = val
return res
原理も分からずに賢い人様が作ったコードをそのまま使うなんて不安!というそこの君、安心したまえ。関数の根幹である "Descriptors._descList" について少し解説していこか。量がめっちゃ多いから、先頭の5つだけ抜き出すな。
Descriptors._descList[:5]
[('MaxEStateIndex',
<function rdkit.Chem.EState.EState.MaxEStateIndex(mol, force=1)>),
('MinEStateIndex',
<function rdkit.Chem.EState.EState.MinEStateIndex(mol, force=1)>),
('MaxAbsEStateIndex',
<function rdkit.Chem.EState.EState.MaxAbsEStateIndex(mol, force=1)>),
('MinAbsEStateIndex',
<function rdkit.Chem.EState.EState.MinAbsEStateIndex(mol, force=1)>),
('qed',
<function rdkit.Chem.QED.qed(mol, w=QEDproperties(MW=0.66, ALOGP=0.46, HBA=0.05, HBD=0.61, PSA=0.06, ROTB=0.65, AROM=0.48, ALERTS=0.95), qedProperties=None)>)]
こんな感じで「記述子」と「記述子を求める関数」がタプル形式で保存されてるんやわ。
はい、ここからがこのコードの最重要部分やで! smilesをmol形式に変換して、先ほど定義したgetMolDescriptors関数を使って一気にバーッと記述子生成していくで。
# SMILESをmol形式に変換しておく
mols = [Chem.MolFromSmiles(i) for i in data["SMILES"]]
# 各分子の記述子を計算
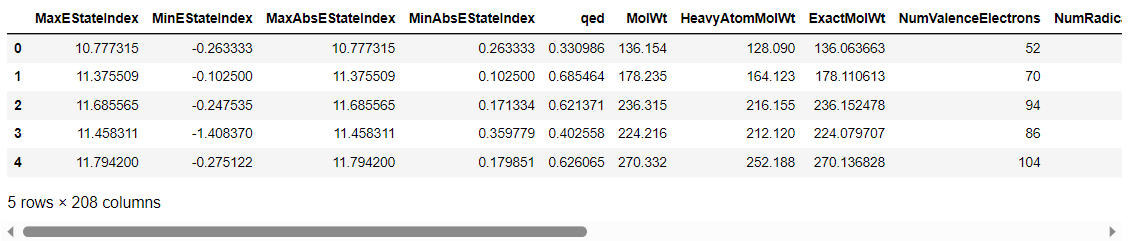
allDescrs = [getMolDescriptors(m) for m in mols]
# 記述子が格納されたデータフレームを生成
df_descriptor = pd.DataFrame(allDescrs)
df_descriptor.head()
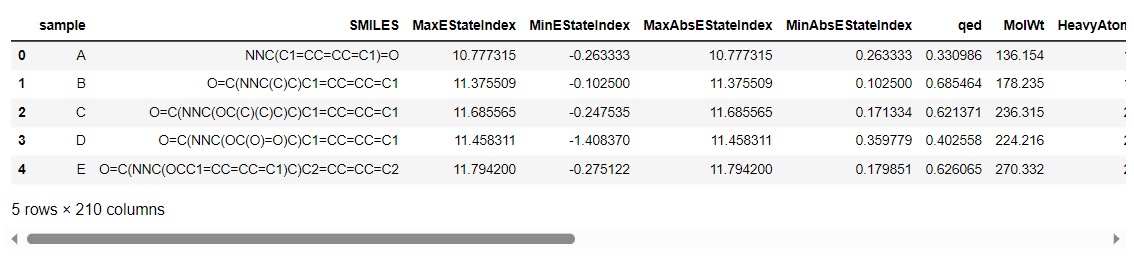
# 読み込んだデータフレームと記述子が格納されたデータフレームを横方向に連結
df = pd.concat([data, df_descriptor], axis=1)
df.head()
3次元記述子の追加
さっき算出した記述子は分子の2次元情報を反映した記述子がメインや。ただ、分子は3次元構造を持っとるので3次元情報も重要や。そこで今回はPBFっちゅう3次元構造に基づく記述子の生成方法を紹介するな。PBFが何かって?簡単に言うたら分子が平面からどんだけ逸脱して立体的かってことを表現するものやけど、詳しいことはこのサイトに分かりやすく書いてあるわ。
ほな、PBF求めていくな。ポイントはRDKitで分子を生成すると毎回立体構造の異なった分子が生成される点やな。つまり計算するたびに毎回異なるPBFが算出されるねん。だから下のコードでは15回計算して平均値を採用するという工夫をしてるねん。ここで昔のアホな俺が質問してるねんけど、優しいおっちゃんが丁寧に答えはってくれて理解深まったわ。様々な配座を取れるような分子はPBFのばらつきが大きいけど、だいたい15回も計算して平均取ったらええ感じなるやろ。知らんけど。
# PBFの計算結果を
df_PBFs_all = pd.DataFrame()
for i in range(15):
PBFs = []
for j in smiles:
mol = Chem.MolFromSmiles(j) # SMILESをMolに変換
molh = Chem.AddHs(mol)
AllChem.EmbedMolecule(molh)
PBF = rdMolDescriptors.CalcPBF(molh)
PBFs.append(PBF)
df_PBFs = pd.DataFrame(PBFs)
df_PBFs_all = pd.concat([df_PBFs_all, df_PBFs], axis=1)
df_PBFs_all.columns = ["PBF1", "PBF2", "PBF3", "PBF4", "PBF5", "PBF6", "PBF7", "PBF8", "PBF9", "PBF10", "PBF11", "PBF12", "PBF13", "PBF14", "PBF15"]
# 各サンプル計算したPBFの平均値を求めておく
df_PBFs_mean = pd.DataFrame(df_PBFs_all.mean(axis=1))
df_PBFs_mean.columns = ["PBF_mean"]
df_PBFs_mean.head()
# PBFの平均値を記述子が格納されたデータフレームの最後に連結する
df = pd.concat([df, df_PBFs_mean], axis=1)
df
# 生成した記述子を含むデータセットをcsvとして保存する
df.to_csv("descriptor.csv", index=False) # index=Falseで列名を除く
PBF以外にもオリジナリティあふれる記述子を生成したいっちゅうんやったら、ここでも参考に地道に実装しなはれ。
最後に
このコード使いこなせたら、いいことRDKit!(いいことあるで!きっと!)