シングルセル解析の概要
シングルセル解析は、細胞一つ一つを個別に調べることにより、生物学的な多様性と複雑性を理解するための強力な手段です。従来の分析では、細胞集団の平均的な特性しか捉えられず、細胞間の微妙な違いを区別できないところを、シングルセル解析ならば細胞の個性を詳細に研究することが可能になります。これにより、健康と病気のプロセスを新たな視点から理解し、より効果的な治療法の開発につながる可能性が広がっています。
環境、用いたデータ
-
環境
python3.12.2
google colaboratory
コマンドライン引数に与えるユーザー名は適宜読み替えてください。 -
データ
GSE219130
また、このデータを用いた元論文の論理構成は以下のようになっています。

今回使用するライブラリは、scanpy, decouplerであり、これらはデータとして10Xgenomicsを用いることとなっているために、10Xgenomicsのデータフォーマットに基づいているデータを選択しました。データはGSE219130にあります。
具体的なコードはgoogle colaboratoryにあり、コードの説明等はこちらでしています。
全体の流れ
シングルセル解析
- データのダウンロードとディレクトリ作成:データを指定されている形にします
- 前処理:データをフィルタリングします
- アノテーション:各細胞がどの細胞型なのか決定します
- パスウェイ解析:代謝経路、シグナル伝達系、タンパク質間相互作用、遺伝子の制御関係等を調べます
- 転写因子の推定:重要な転写因子(遺伝子の発現を制御するタンパク質)を同定します
- 生物学的プロセス推定:各細胞でよく発現している遺伝子を調べます
- 2群の比較:比較して考察します
疑似バルク解析
- データ成形
- 前処理
- 発現差解析(DEG、DEA)
- 転写因子推定
- パスウェイ解析
- 生物学的プロセス推定
まとめ
全体をまとめて考察します
シングルセル解析
データのダウンロードとディレクトリ作成
データの検索方法
GSE219130に入り、
httpからダウンロードします。今回はすべてのデータを使うのでhttpですが、customで必要に応じてダウンロードしてもよいでしょう。
次に10Xgenomicsに基づいてディレクトリを作ります。

この形にします。今回4個体いるために、それぞれのファイルの前に名前をつけることで読み込むことができます。

scanpy.read_10x_mtxのprefix参照
また、google colaboratoryを用いるのでこれをGoogle Driveにアップグレードしています。
なお各個体のmetadataは以下の通りです。
| sample name | genotype | treatment |
|---|---|---|
| WT_CD | wild-type | control diet |
| WT_ID | wild-type | iodine deficient diet |
| APT_CD | mutant APP, PSEN1, Tau overexpressed | control diet |
| APT_ID | mutant APP, PSEN1, Tau overexpressed | iodine deficient diet |
簡潔にまとめると、APTはアルツハイマーでIDはヨウ素が欠乏しているということです。
解析開始
前処理まではscanpyのチュートリアルに基づき、その後はdecopulerに基づいてすすめていきます。
目次は以下です
- 前処理
- アノテーション
- パスウェイ解析
- 転写因子の推定
- 遺伝子マーカー抽出(生物学的用語、分子プロセス推定)
- WD_CDとAPT_IDの比較
前処理
ダウンロードしたデータは4つの個体からなりますが、まずは1個体(APT_ID)の解析を行います。
file_path = "/content/drive/MyDrive/filtered_gene_bc_matrices/hg19/"
adata = sc.read_10x_mtx(
file_path,
var_names='gene_symbols',
cache=True,
prefix="APT_ID_")
adata.var_names_make_unique()
adata
何も処理していない最初の発現の様子はこんなかんじです
sc.pl.highest_expr_genes(adata, n_top=20,)
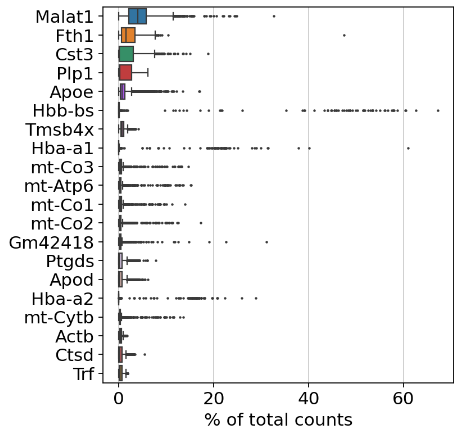
ここからどんどんフィルタリングします。
発現が少なすぎる細胞、遺伝子をフィルタリング
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata
ミトコンドリア遺伝子の割合が高い細胞をフィルタリング
adata = adata[adata.obs.n_genes_by_counts < 7000, :]
adata = adata[adata.obs.pct_counts_mt < 20, :]
adata
HVG (Highly Variable Genes)のフィルタリング
adata = adata[:, adata.var.highly_variable]
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
adata
主成分分析を行います。これはデータの次元を削減して特徴を捉えるというものです。
sc.tl.pca(adata, svd_solver='arpack')
近傍グラフの計算を行います。これは細胞間の距離(類似性や関連性)を数値化し、細胞間の関係をグラフ構造で表現することを目的としています。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
UMAPを使用してグラフを2次元に埋め込み、図示してみます。('Tmem119','Tyrobp','Plpp3')は適当に選んだものです。
sc.pl.umap(adata, color=['Tmem119','Tyrobp','Plpp3'], use_raw=False)
近傍グラフのクラスタリングを行います。
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
n_iterations=2,
directed=False,
)
2次元で図示すると以下のようになりました。

このようにクラスタリングできましたが、各クラスターがどの細胞タイプを表しているのかがわかりません。そこでここかアノテーションを行い、これを推定していきます。
アノテーション
各細胞がどの細胞型なのか決定するものです。
ここからはdecouplerのチュートリアルに基づきます。
マーカー遺伝子は様々な研究で発見され注釈が付けられており、それらを整理しているデータベースが存在します。それらを用いていきます。今回は主にOmniPathを使用します。
データベースをダウンロードします。
markers = dc.get_resource('PanglaoDB')
markers
マウスに変えてフィルタリングします
# Filter by canonical_marker and mouse
markers = markers[markers['mouse'] & markers['canonical_marker'] & (markers['mouse_sensitivity'] > 0.5)]
# Remove duplicated entries
markers = markers[~markers.duplicated(['cell_type', 'genesymbol'])]
markers
Over Representation Analysis (ORA) methodを実行し、これに基づいてアノテーションを行います。
ORAとはデータ内の特定のグループや属性が過剰に表現されているかどうかを判断するものです
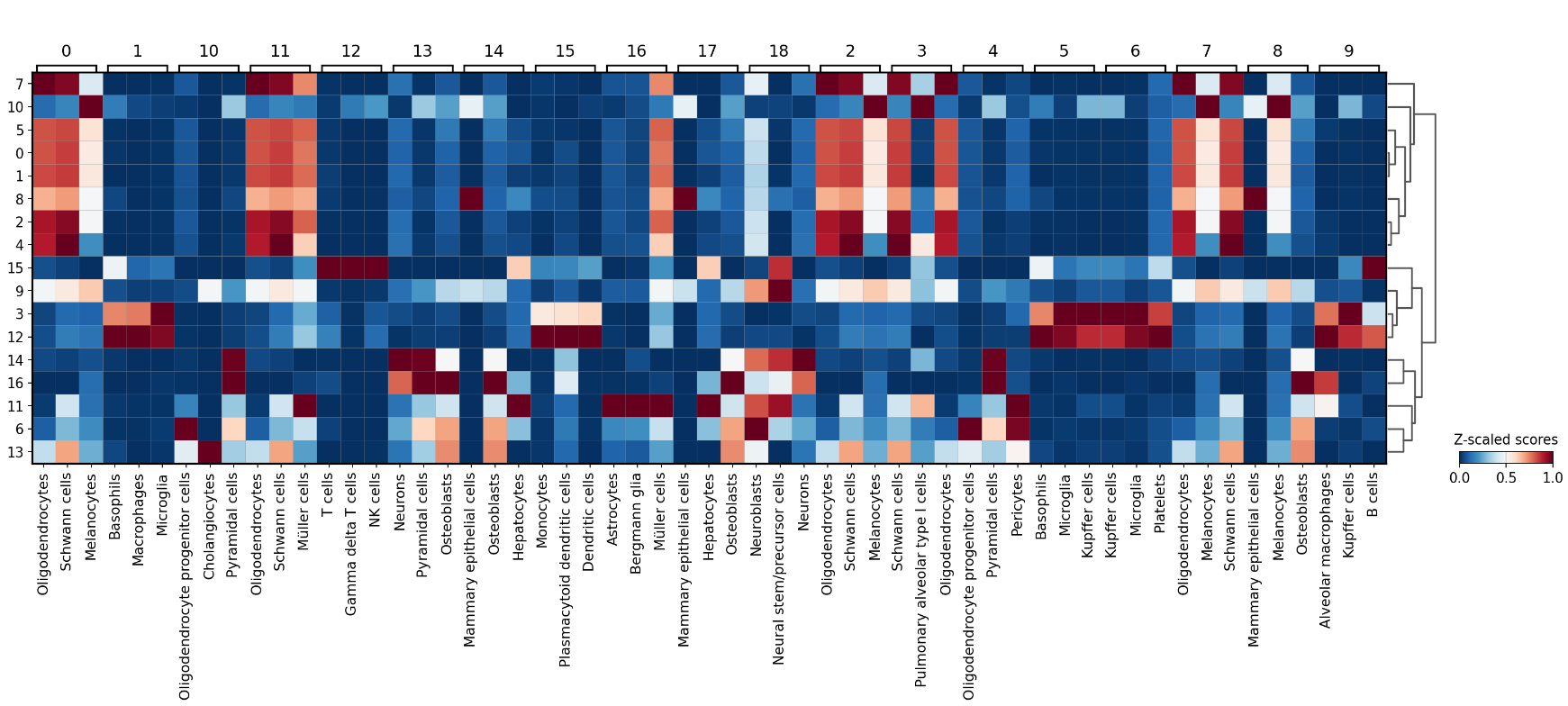
視覚化すると以下のようになります。
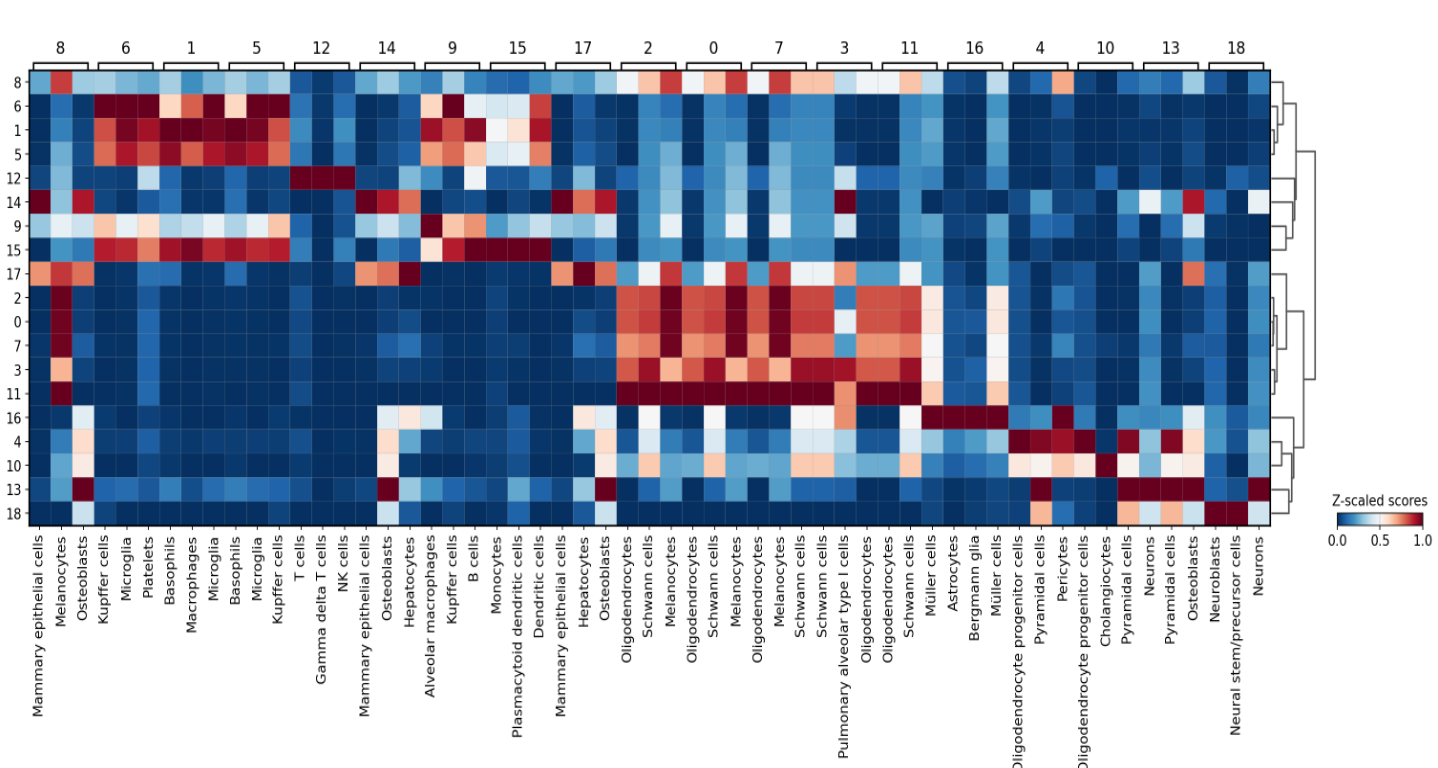
Z scale scoreは発現量を正規化した値です
下の横軸はアノテーションされた細胞、左の縦軸はクラスターの番号を表しています。上の横軸もクラスターを表しています。右のトーナメント表のようなものは樹形図です。
この図が見にくいのは、上の横軸があるためで、まずは下の横軸と左の縦軸に注目しましょう。こうすると、赤に近い色のところは発現量が多いことを表しています。
非常にわかりやすいです。
上の横軸にあるクラスターの上位3つの細胞が下の横軸に書いてあります。
また、樹形図では、似ているクラスターが近くに置かれます。
アノテーションは曖昧さを含みますが、とりあえずデータ上それらしいものを決定します。
annotation_dict = df.groupby('group').head(1).set_index('group')['names'].to_dict()
annotation_dict
結果は以下のようになります。
| Cluster | Cell Type |
|---|---|
| 0 | Oligodendrocytes |
| 1 | Basophils |
| 2 | Oligodendrocytes |
| 3 | Schwann cells |
| 4 | Oligodendrocyte progenitor cells |
| 5 | Basophils |
| 6 | Kupffer cells |
| 7 | Oligodendrocytes |
| 8 | Mammary epithelial cells |
| 9 | Alveolar macrophages |
| 10 | Oligodendrocyte progenitor cells |
| 11 | Oligodendrocytes |
| 12 | T cells |
| 13 | Neurons |
| 14 | Mammary epithelial cells |
| 15 | Monocytes |
| 16 | Astrocytes |
| 17 | Mammary epithelial cells |
| 18 | Neuroblasts |
近傍グラフに注釈をつけます。

パスウェイ解析
パスウェイとは、遺伝子やタンパク質や化合物等の相互作用を表現したものです。代謝経路やシグナル伝達系やタンパク質間相互作用、遺伝子の制御関係等の情報を含みます。パスウェイ解析を行うと、ある遺伝子のリストについて、遺伝子全体と比較して有意に多く観測されるパスウェイを検出できます。
PROGENyはパスウェイとその標的遺伝子の厳選されたコレクションと各相互作用の重みを含む包括的なリソースであり、これを用います。
データベースをダウンロードします。
progeny = dc.get_progeny(organism='Mus musculus', top=500)
progeny
multivariate linear model(MLM) によるアクティビティ推論を行い視覚化します。
MLMは複数の目的変数を同時に予測するための線形モデルです。このモデルの特徴は複数の目的変数を扱うことです。また、変数間の相関関係も考慮にいれます。
説明変数は各パスウェイにおける遺伝子の相互作用の重みを表します。目的変数は遺伝子発現量です。
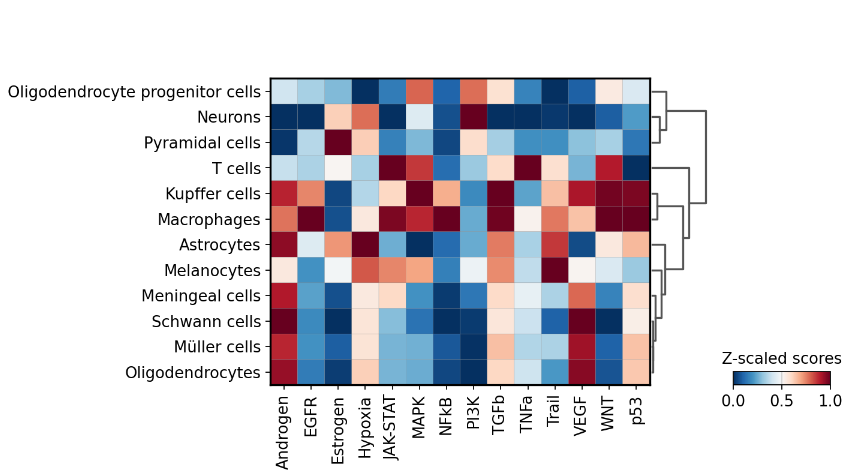
ヒートマップを図示します。

横軸はパスウェイ、縦軸は細胞を表しています。Z-scoreが1に近いほど関係性が高いことを表しています。右の樹形図では近くにあるクラスターが類似していることを表しています。
各経路の説明
Androgen : 男性の生殖器官の成長と発達に関与します。
EGFR : 哺乳類細胞の成長、生存、遊走、アポトーシス、増殖、分化を調節します。
Estrogen: 女性の生殖器の成長と発達を促進します。
Hypoxia: O2 レベルが低い場合、血管新生と代謝の再プログラミングを促進します。
JAK-STAT : 免疫、細胞分裂、細胞死、腫瘍形成に関与します。
MAPK : 外部シグナルを統合し、細胞の成長と増殖を促進します。
NFkB : 免疫応答、サイトカイン産生、細胞生存を調節します。
p53 : 細胞周期、アポトーシス、DNA修復、腫瘍抑制を制御します。
PI3K : 成長と増殖を促進します。
TGFb : ほとんどの組織の発生、恒常性、修復に関与します。
TNFa : 造血、免疫監視、腫瘍退縮、感染からの保護を仲介します。
Trail : アポトーシスを誘導します。
VEGF : 血管新生、血管透過性、細胞遊走を媒介します。
WNT : 発生および組織修復中の器官の形態形成を調節します。
参照
転写因子活性の推定
計測が容易な遺伝子発現の変化から転写因子(遺伝子の発現を制御するタンパク質)の状態を予測し、重要な転写因子を同定することができます。
CollecTRIを用います。
データベースをダウンロードします。
net = dc.get_collectri(organism='Mus musculus', split_complexes=False)
net
univariate linear model(ULM)による推論を行います。
ULMでは1つの目的変数を予測するための線形モデルです。このモデルの特徴は変数間の相関関係も考慮にいれることです。
説明変数は各パスウェイにおける遺伝子の相互作用の重みを表します。目的変数は遺伝子発現量です。
細胞タイプごとに上位の転写因子を特定します。
n_markers = 3
source_markers = df.groupby('group').head(n_markers).groupby('group')['names'].apply(lambda x: list(x)).to_dict()
source_markers
細胞と上位の転写因子の対応を図にします。
| Cell Type | Transcriptional Activator |
|---|---|
| Astrocytes | Nkx3-2, Lrrfip1, Tbx18 |
| Basophils | Spi1, Irf1, Elf1 |
| Kupffer cells | Klf2, Myod1, Rarg |
| Mammary epithelial cells | Atoh7, Pitx1, Hdac1 |
| Monocytes | Stat2, Irf3, Irf1 |
| Neuroblasts | Sox11, Dlx2, Foxj1 |
| Neurons | Tbr1, Atoh7, Znf263 |
| Oligodendrocyte progenitor cells | Barx2, Nkx6-1, Gli2 |
| Oligodendrocytes | Myt1, Nkx2-2, Fev |
| Schwann cells | Nkx2-2, Hoxc6, Pura |
| T cells | Hivep2, Zfx, Pax8 |
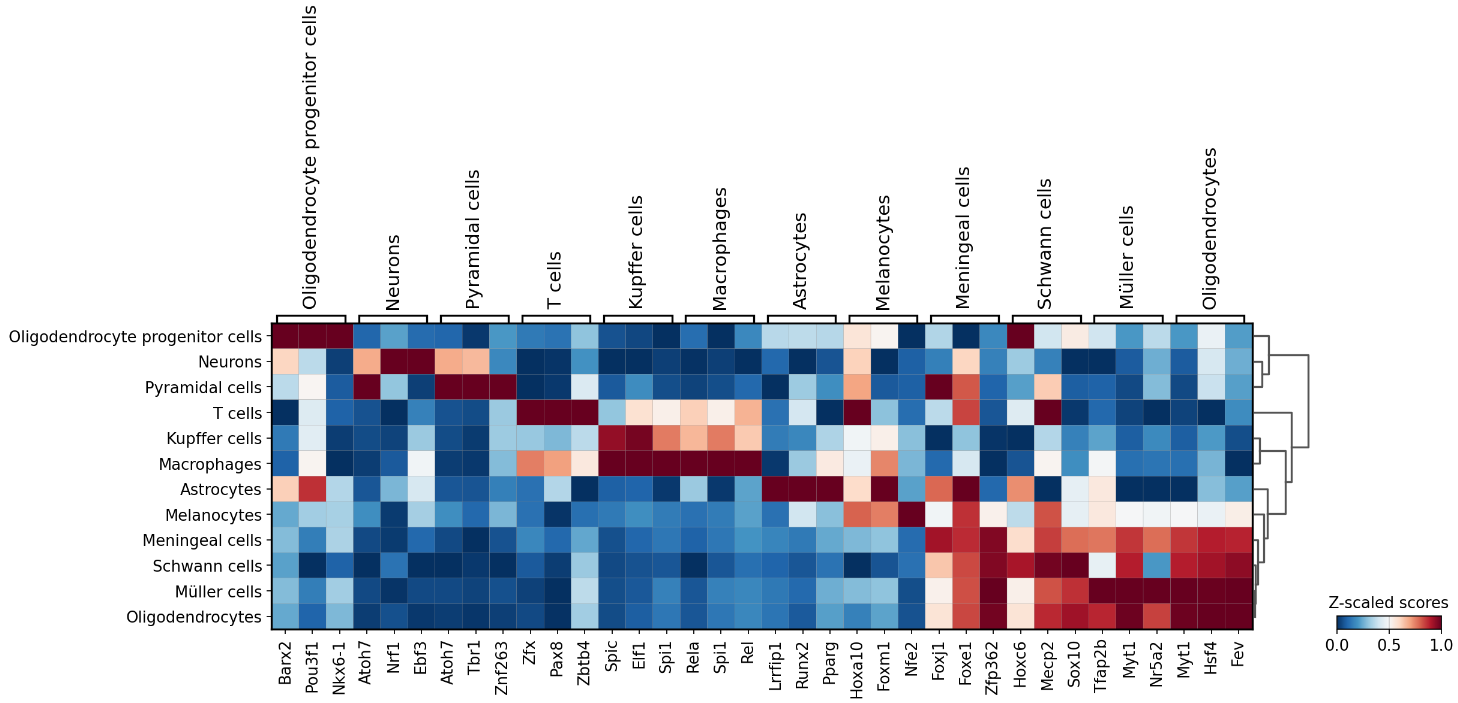
ヒートマップは以下です。
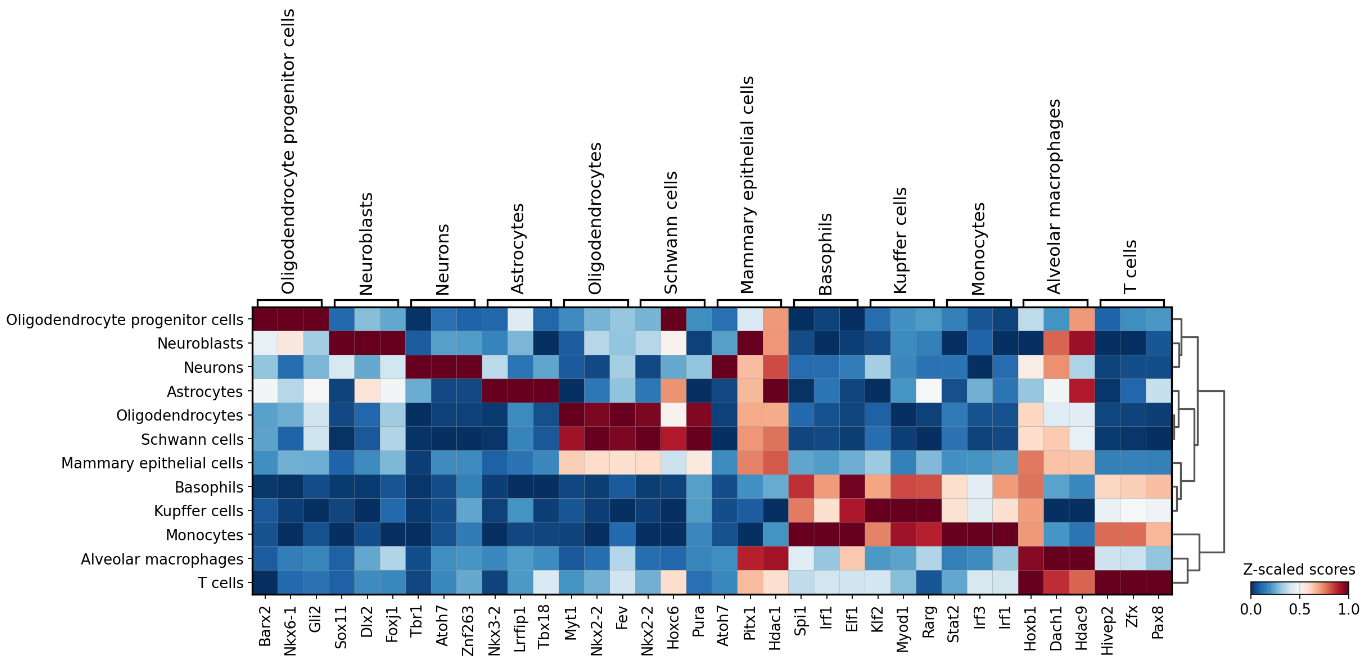
下の軸は転写因子(タンパク質)を表します。左の軸は細胞、上の軸も細胞です。右は樹形図を表します。
生物学的プロセス推定
Gene Setを推定します。
MSigDBを用います。これはさまざまな生物学的プロセスに注釈が付けられた遺伝子セットのコレクションを含むリソースです。
msigdb = dc.get_resource('MSigDB')
msigdb
ヒトからマウスに変えたり、データをきれいにした後Representation Analysis (ORA)を実行します。
細胞と各細胞での上位の遺伝子セットを表にします。HALLMARKはMSigDBでの遺伝子セットの先頭につけられている名前です。
| Cell Type | Genes Sets |
|---|---|
| Alveolar macrophages | HALLMARK_DNA_REPAIR, HALLMARK_PEROXISOME, HALLMARK_XENOBIOTIC_METABOLISM |
| Astrocytes | HALLMARK_BILE_ACID_METABOLISM, HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION, HALLMARK_UV_RESPONSE_DN |
| Basophils | HALLMARK_COMPLEMENT, HALLMARK_TNFA_SIGNALING_VIA_NFKB, HALLMARK_ALLOGRAFT_REJECTION |
| Kupffer cells | HALLMARK_COMPLEMENT, HALLMARK_P53_PATHWAY, HALLMARK_TNFA_SIGNALING_VIA_NFKB |
| Mammary epithelial cells | HALLMARK_APICAL_SURFACE, HALLMARK_HEME_METABOLISM, HALLMARK_SPERMATOGENESIS |
| Monocytes | HALLMARK_INTERFERON_GAMMA_RESPONSE, HALLMARK_INTERFERON_ALPHA_RESPONSE, HALLMARK_INFLAMMATORY_RESPONSE |
| Neuroblasts | HALLMARK_G2M_CHECKPOINT, HALLMARK_MYC_TARGETS_V1, HALLMARK_E2F_TARGETS |
| Neurons | HALLMARK_SPERMATOGENESIS, HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| Oligodendrocyte progenitor cells | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_SPERMATOGENESIS, HALLMARK_NOTCH_SIGNALING |
| Oligodendrocytes | HALLMARK_MYOGENESIS, HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY, HALLMARK_ADIPOGENESIS |
| Schwann cells | HALLMARK_APICAL_SURFACE, HALLMARK_MYOGENESIS, HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| T cells | HALLMARK_ALLOGRAFT_REJECTION, HALLMARK_INTERFERON_ALPHA_RESPONSE, HALLMARK_INTERFERON_GAMMA_RESPONSE |
ヒートマップを図示します。ここではHALLMARK_があると図が見にくくなるのでこれを除いています。

下の軸は遺伝子セット、左軸は細胞、上軸も細胞、右軸は樹形図です。
WD_CDとAPT_IDの比較
改めて各個体がなんであるかを示します。
| sample name | genotype | treatment |
|---|---|---|
| WT_CD | wild-type | control diet |
| WT_ID | wild-type | iodine deficient diet |
| APT_CD | mutant APP, PSEN1, Tau overexpressed | control diet |
| APT_ID | mutant APP, PSEN1, Tau overexpressed | iodine deficient diet |
つまりAPT_IDはアルツハイマーでヨウ素欠乏のマウスです。
ここでは、これまでAPT_IDで行ったことをWD_CDでも行い、違いを考察します。
WD_CDとAPT_IDの比較をおこなう理由は以下の通りです。
このデータで元論文が言いたいことは
We demonstrate that microglial ecto-5’-nucleotidase (CD73) is reduced in the hypothyroid state and that its inhibition contributes to immune tolerance in microglia.
つまりAPT_IDではミクログリア、CD73(NT5E)が減少している発現量が低下していることを示したい
また以下も参考にする。
CD73/アデノシン経路は、Treg細胞の強力な追加抑制経路であり
5′-nucleotidase (5′-NT), also known as ecto-5′-nucleotidase or CD73 (cluster of differentiation 73), is an enzyme that in humans is encoded by the NT5E gene.[5] CD73 commonly serves to convert AMP to adenosine.
ミクログリアのマーカー
TMEM119| CD11bとCD45| Iba1| CX3CR1| F4/80 | CD68 | CD40
ectonucleoside triphosphate diphosphohydrolase1(CD39)とecto-5′-nucleotidase
(CD73)によって脱リン酸化され,細胞外アデノシンへと
変換される
まとめ:APT_IDでは免疫寛容がおこりミクログリア、CD73(NT5E)、アデノシンが減少し、Treg細胞亢進、TMEM119, CD11b,CD45, Iba1, CX3CR1, F4/80, CD68, CD40が減少しているはず。
最もしたい比較はWD_CDとAPT_IDであり、以上をふまえながら比較していきます。
比較するものは以下の通りです。
- アノテーション
- パスウェイ解析
- 転写因子活性
- 生物学的用語、分子プロセス
アノテーション
各細胞がどの細胞型なのか決定するものです。
ヒートマップは以下。
WD_CD
APT_ID
詳しすぎて全体を見てもピンとこない。見たいものが決まっている時に効果を発揮すると思われる。(改めて人力でチェックしながらアノテーションする時など)
順にWD_CD, APT_ID
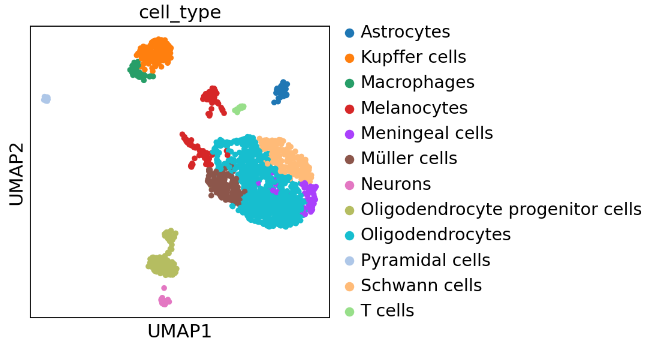
これは次元削減手法のUMAP(Uniform Manifold Approximation and Projection)によって生成された図です。UMAPは高次元データを2次元や3次元の低次元空間に写像する手法で、類似したデータ点が近くにまとまるように配置されます。ここでは類似した点が細胞としてまとまっています。ただし座標の絶対値そのものには直接的な意味はありません。位置関係から類似性を判断するのが目的です。つまり左の図と右の図の位置を比較することに意味はなく、各図の中での位置関係を考えることに意味があるということです。
クラスターとそれに対応する細胞の比較をします。
| Cluster | Cell Type (WD_CD) | Cell Type (APT_ID) |
|---|---|---|
| 0 | Oligodendrocytes | Oligodendrocytes |
| 1 | Oligodendrocytes | Basophils |
| 2 | Oligodendrocytes | Oligodendrocytes |
| 3 | Kupffer cells | Schwann cells |
| 4 | Schwann cells | Oligodendrocyte progenitor cells |
| 5 | Müller cells | Basophils |
| 6 | Oligodendrocyte progenitor cells | Kupffer cells |
| 7 | Oligodendrocytes | Oligodendrocytes |
| 8 | Meningeal cells | Mammary epithelial cells |
| 9 | Melanocytes | Alveolar macrophages |
| 10 | Melanocytes | Oligodendrocyte progenitor cells |
| 11 | Astrocytes | Oligodendrocytes |
| 12 | Macrophages | T cells |
| 13 | Oligodendrocyte progenitor cells | Neurons |
| 14 | Neurons | Mammary epithelial cells |
| 15 | T cells | Monocytes |
| 16 | Pyramidal cells | Astrocytes |
| 17 | Mammary epithelial cells | |
| 18 | Neuroblasts |
ここにミクログリアは登場しない。。。
なぜミクログリアなのかというと、前に述べたがAPT_IDではミクログリア、CD73(NT5E)が減少している発現量が低下していることを示したいという事情によります。
アノテーションは最終的に人の手で行う必要があり、そこに今回の限界があったのではないかと考えます。つまりどれかがミクログリアであってほしかった。
パスウェイ解析
ヒートマップは以下。
WD_CD
APT_ID

全体を概観してもよくわからない。
しかしながら例えばEGFRに注目するとWD_CDでKupper cellとMacrophagesという血管透過性を必要とする細胞で発現しているのに対し、APT_IDではBasophils(好塩基球),Kupper cell, Monocytesで発現しており何らかの免疫反応が亢進していることがうかがえる。ここから非常に雑だが、免疫寛容の方向を向いている要素はあると言えるのではないだろうか。
JAK-STATに注目してみるとWD_CDではMacrophagesとT cellでよく発現し、APT_IDではMonocytesでよく発現している。ここからは特に何も言えない。
JAK-STATにさらに注目してみる。
順にWD_CD, APT_ID


JAK-STATはAPT_IDのMonocytesでよく発現している。WD_CDのMacrophagesとT cellよりも強く発現していることがわかる。ここからWD_CDとAPT_IDではAPT_IDのほうでJAK-STATがよく働いているといえるのではないか。JAK-STATは雑にいうと免疫を強くする作用がある。よって免疫寛容の方向を向く要素がもう一つ増えた。
転写因子活性
計測が容易な遺伝子発現の変化から転写因子(遺伝子の発現を制御するタンパク質)の状態を予測し、重要な転写因子を同定することができます。
ヒートマップは以下です。
WD_CD
APT_ID

これもまた、全体を見ようとすると詳しすぎる。目が眩んでくる。
細胞と転写因子の対応についての表は以下。
| Cell Type | Transcriptional Activator (WD_CD) | Transcriptional Activators (APT_ID) |
|---|---|---|
| Astrocytes | Lrrfip1, Runx2, Pparg | Nkx3-2, Lrrfip1, Tbx18 |
| Basophils | Spi1, Irf1, Elf1 | |
| Kupffer cells | Spic, Elf1, Spi1 | Klf2, Myod1, Rarg |
| Mammary epithelial cells | Atoh7, Pitx1, Hdac1 | |
| Monocytes | Stat2, Irf3, Irf1 | |
| Neuroblasts | Sox11, Dlx2, Foxj1 | |
| Neurons | Atoh7, Nrf1, Ebf3 | Tbr1, Atoh7, Znf263 |
| Oligodendrocyte progenitor cells | Barx2, Pou3f1, Nkx6-1 | Barx2, Nkx6-1, Gli2 |
| Oligodendrocytes | Myt1, Hsf4, Fev | Myt1, Nkx2-2, Fev |
| Schwann cells | Hoxc6, Mecp2, Sox10 | Nkx2-2, Hoxc6, Pura |
| T cells | Zfx, Pax8, Zbtb4 | Hivep2, Zfx, Pax8 |
| Macrophages | Rela, Spi1, Rel | |
| Melanocytes | Hoxa10, Foxm1, Nfe2 | |
| Meningeal cells | Foxj1, Foxe1, Zfp362 | |
| Müller cells | Tfap2b, Myt1, Nr5a2 | |
| Pyramidal cells | Atoh7, Tbr1, Znf263 |
免疫反応の中心であるT cellsに注目してみる。 WD_CDではZfx, Pax8, Zbtb4。APT_IDではHivep2, Zfx, Pax8である。
Zfx
Probable transcriptional activator
普通に転写活性化させてるのかな、、、
Pax8
This nuclear protein is involved in thyroid follicular cell development and expression of thyroid-specific genes. Mutations in this gene have been associated with thyroid dysgenesis, thyroid follicular carcinomas and atypical follicular thyroid adenomas.
甲状腺において必要な遺伝子を発現させる。これがWD_CDだけに発現していたらきれいだった。
Zbtb4
Involved in cellular response to DNA damage stimulus and negative regulation of transcription by RNA polymerase II
普通に転写を負に制御してる感じ
Hivep2
It may act in T-cell activation
T細胞の活性化に寄与している可能性がある。
まとめ:ここに今回の結論を裏付けるものはないと思われる。
上で見たPax8について詳しく比較してみる。
順にWD_CD, APT_ID

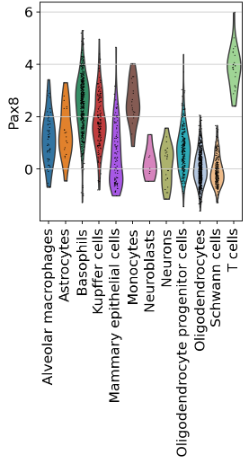
T cellでよく発現しているのは当然だが、そのほかではそこまで差はないように思える。しいて言えばWD_CDのMacrohagesでよく発現している。しかし示したいことを裏付けるものではない。
生物学的プロセス
Gene Setを推定していきます。例によってHALLMARK_は心の目で見てください。
WD_CD
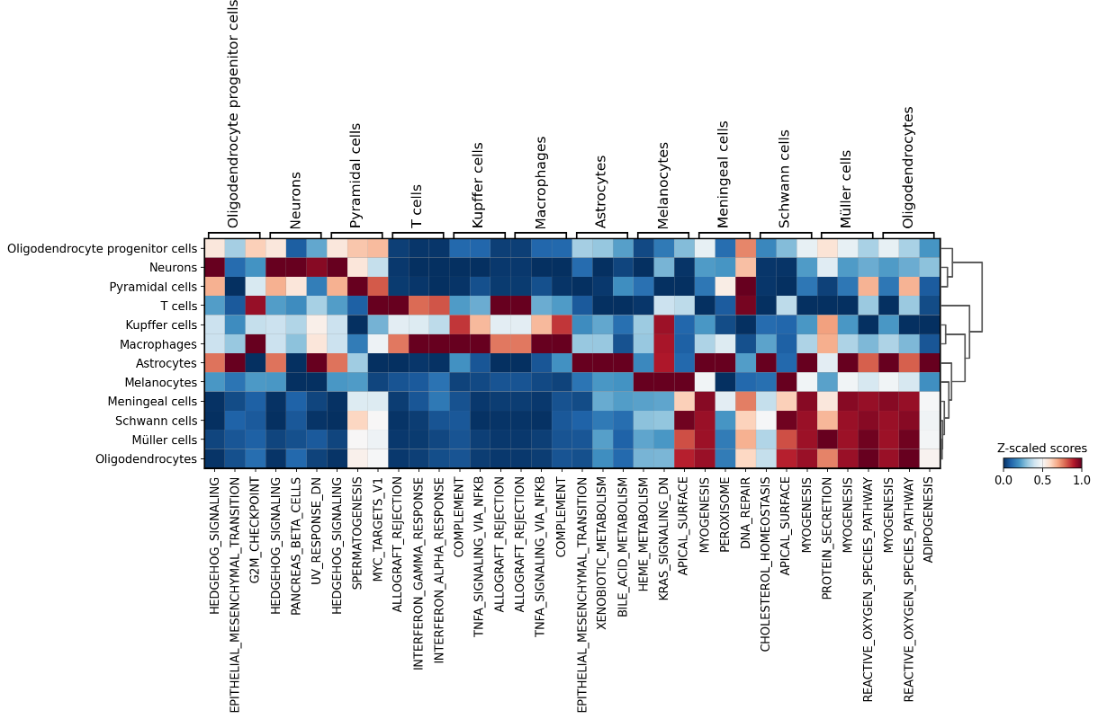
APT_ID
細胞と遺伝子セットの関係を表で表します。
| Cell Type | Genes sets(WD_CD) | Genes sets(APT_ID) |
|---|---|---|
| Alveolar macrophages | HALLMARK_DNA_REPAIR, HALLMARK_PEROXISOME, HALLMARK_XENOBIOTIC_METABOLISM | |
| Astrocytes | HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION, HALLMARK_XENOBIOTIC_METABOLISM, HALLMARK_BILE_ACID_METABOLISM | HALLMARK_BILE_ACID_METABOLISM, HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION, HALLMARK_UV_RESPONSE_DN |
| Basophils | HALLMARK_COMPLEMENT, HALLMARK_TNFA_SIGNALING_VIA_NFKB, HALLMARK_ALLOGRAFT_REJECTION | |
| Kupffer cells | HALLMARK_COMPLEMENT, HALLMARK_TNFA_SIGNALING_VIA_NFKB, HALLMARK_ALLOGRAFT_REJECTION | HALLMARK_COMPLEMENT, HALLMARK_P53_PATHWAY, HALLMARK_TNFA_SIGNALING_VIA_NFKB |
| Mammary epithelial cells | HALLMARK_APICAL_SURFACE, HALLMARK_HEME_METABOLISM, HALLMARK_SPERMATOGENESIS | |
| Monocytes | HALLMARK_INTERFERON_GAMMA_RESPONSE, HALLMARK_INTERFERON_ALPHA_RESPONSE, HALLMARK_INFLAMMATORY_RESPONSE | |
| Neuroblasts | HALLMARK_G2M_CHECKPOINT, HALLMARK_MYC_TARGETS_V1, HALLMARK_E2F_TARGETS | |
| Neurons | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_PANCREAS_BETA_CELLS, HALLMARK_UV_RESPONSE_DN | HALLMARK_SPERMATOGENESIS, HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| Oligodendrocyte progenitor cells | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION, HALLMARK_G2M_CHECKPOINT | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_SPERMATOGENESIS, HALLMARK_NOTCH_SIGNALING |
| Oligodendrocytes | HALLMARK_MYOGENESIS, HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY, HALLMARK_ADIPOGENESIS | HALLMARK_MYOGENESIS, HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY, HALLMARK_ADIPOGENESIS |
| Schwann cells | HALLMARK_CHOLESTEROL_HOMEOSTASIS, HALLMARK_APICAL_SURFACE, HALLMARK_MYOGENESIS | HALLMARK_APICAL_SURFACE, HALLMARK_MYOGENESIS, HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| T cells | HALLMARK_ALLOGRAFT_REJECTION, HALLMARK_INTERFERON_GAMMA_RESPONSE, HALLMARK_INTERFERON_ALPHA_RESPONSE | HALLMARK_ALLOGRAFT_REJECTION, HALLMARK_INTERFERON_GAMMA_RESPONSE, HALLMARK_INTERFERON_ALPHA_RESPONSE |
| Macrophages | HALLMARK_ALLOGRAFT_REJECTION, HALLMARK_TNFA_SIGNALING_VIA_NFKB, HALLMARK_COMPLEMENT | |
| Melanocytes | HALLMARK_HEME_METABOLISM, HALLMARK_KRAS_SIGNALING_DN, HALLMARK_APICAL_SURFACE | |
| Meningeal cells | HALLMARK_MYOGENESIS, HALLMARK_PEROXISOME, HALLMARK_DNA_REPAIR | |
| Müller cells | HALLMARK_PROTEIN_SECRETION, HALLMARK_MYOGENESIS, HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY | |
| Pyramidal cells | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_SPERMATOGENESIS, HALLMARK_MYC_TARGETS_V1 |
Neuronsに注目してみよう。
| Cell Type | Marker Genes(WD_CD) | Marker Genes(APT_ID) |
|---|---|---|
| Neurons | HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_PANCREAS_BETA_CELLS, HALLMARK_UV_RESPONSE_DN | HALLMARK_SPERMATOGENESIS, HALLMARK_HEDGEHOG_SIGNALING, HALLMARK_OXIDATIVE_PHOSPHORYLATION |
共通するのがHALLMARK_HEDGEHOG_SIGNALING、WD_CDのみなのがHALLMARK_PANCREAS_BETA_CELLS, HALLMARK_UV_RESPONSE_DN、APT_IDのみなのがHALLMARK_SPERMATOGENESIS,HALLMARK_OXIDATIVE_PHOSPHORYLATIONです。
HALLMARK_HEDGEHOG_SIGNALING (ヘッジホッグシグナル伝達に関与する遺伝子群)
HALLMARK_PANCREAS_BETA_CELLS (膵臓のβ細胞で特異的に発現上昇している遺伝子群)
HALLMARK_UV_RESPONSE_DN (紫外線照射により発現低下する遺伝子群)
HALLMARK_SPERMATOGENESIS (精子形成過程で発現上昇する遺伝子群)
HALLMARK_OXIDATIVE_PHOSPHORYLATION (酸化的リン酸化に関与するタンパク質をコードする遺伝子群)
正直どれも微妙、、
両者ともによく発現していたHEDGEHOG_SIGNALINGを詳しく見てみる。
順にWD_CD, APT_ID

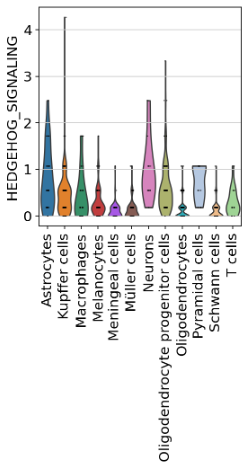
Neuronsの中では上位だが、ほかの細胞と比べた時Neurons自体の発現が特に多いというわけではなかった。
疑似バルク解析
疑似バルク解析は任意の集団で比較することができます。バルク解析では個体、あるいは臓器ごとの比較しかできませんが、疑似バルク解析では明確な細胞同一性クラスターがある場合、それらを一つの集団として見立てて互いに比較することができます。
目次は以下です。
- データ成形
- 前処理
- 発現差解析(DEG、DEA)
- 転写因子推定
- パスウェイ解析
- 生物学的用語、プロセス推定
データ形成
ここで用いるデータは生カウントなので、最初にダウンロードしたデータをデータフレームの形にします。そののちデータフレームをまとめてanndataを作成します。
#データフレームを読み込む
df1 = pd.read_csv('/content/drive/MyDrive/single cell RNA-seq_write/WT_CD.tsv', sep='\t', index_col=0)
df2 = pd.read_csv('/content/drive/MyDrive/single cell RNA-seq_write/WT_ID.tsv', sep='\t', index_col=0)
df3 = pd.read_csv('/content/drive/MyDrive/single cell RNA-seq_write/APT_CD.tsv', sep='\t', index_col=0)
df4 = pd.read_csv('/content/drive/MyDrive/single cell RNA-seq_write/APT_ID.tsv', sep='\t', index_col=0)
ここからデータを成形していってadataを作成します。
adata = pd.concat([df1_sum, df2_sum, df3_sum, df4_sum], axis=0).fillna(0)
adata
| Gm19938 | Rgs20 | Vxn | ... | Tspyl2 | Nhs | Tceanc | |
|---|---|---|---|---|---|---|---|
| WD_CD | 250.0 | 69.0 | 367.0 | ... | 0.0 | 0.0 | 0.0 |
| WD_ID | 252.0 | 0.0 | 351.0 | ... | 0.0 | 0.0 | 0.0 |
| APT_CD | 0.0 | 82.0 | 437.0 | ... | 0.0 | 0.0 | 0.0 |
| APT_ID | 0.0 | 100.0 | 568.0 | ... | 572.0 | 34.0 | 128.0 |
次にadata.obsを作成する必要がありますが、WD_CDとAPT_IDの2つの比較はできません。2つの状態の比較をしたいなら4つ以上のサンプルが必要らしいです。(エラーがでます)そこでWD_CD, WD_ID, APT_CDをcondition(wild), APT_IDをtreatmentとして比較をおこないます。ここでWD_CDをcontrolとしてAPT_IDをtreatmentとすることに異論はないと思いますが、WD_ID, APT_CDの扱いには疑問が残ります。
# データフレームの作成
df = pd.DataFrame({'condition': ['control','control','control', 'treatment']}, index=['WD_CD','WD_ID','APT_CD', 'APT_ID'])
adata.obs = df
adata.obs
| condition | |
|---|---|
| WD_CD | control |
| WD_ID | control |
| APT_CD | control |
| APT_ID | treatment |
前処理
発現量の少なすぎる遺伝子、細胞をフィルタリングします。異なるカウントの合計とサンプル数で遺伝子の発現頻度を観察できます。破線は現在の閾値を示し、右上隅の遺伝子のみが保持されることを意味します。
dc.plot_filter_by_expr(adata, group='condition', min_count=35, min_total_count= 50, large_n=1, min_prop=1)
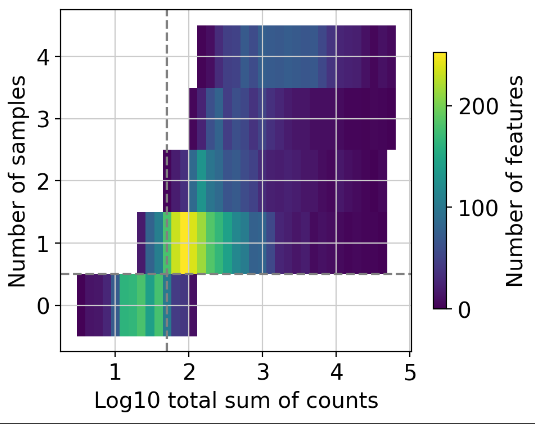
フィルタリングします。
# Obtain genes that pass the thresholds
genes = dc.filter_by_expr(adata, group='condition', min_count=35, min_total_count=50, large_n=1, min_prop=1)
# Filter by these genes
adata = adata[:, genes].copy()
adata
発現差解析(DEA,DEG)
発現差解析についてはQuitaを参考にしてください。
ここではPyDESeq2を用います。
# Build DESeq2 object
inference = DefaultInference(n_cpus=8)
dds = DeseqDataSet(
adata=adata,
design_factors='condition',
refit_cooks=True,
inference=inference,
)
# Compute LFCs
dds.deseq2()
# Extract contrast
stat_res = DeseqStats(
dds,
contrast=["condition", 'treatment', 'control'],
inference=inference
)
# Compute Wald test
stat_res.summary()
結果は以下のようになります。
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| Gm19938 | 151.1 | -10.3 | 5.3 | -1.9 | 0.053 | 0.4 |
| Rgs20 | 59.3 | 0.8 | 4.0 | 0.2 | 0.842 | 0.999 |
| Vxn | 424.3 | 0.3 | 3.0 | 0.1 | 0.917 | 0.999 |
| Csp1 | 197.9 | -13.6 | 9.3 | -1.4 | 0.157 | 0.5 |
| Prex2 | 83.1 | -1.2 | 3.3 | -0.4 | 0.720 | 0.999 |
| ... | ... | ... | ... | ... | ... | ... |
| Gprasp2 | 65.7 | 10.4 | 4.8 | 1.5 | 0.031 | 0.386 |
| Wnk3 | 34.7 | 9.5 | 5.3 | 1.8 | 0.073 | 0.470 |
| Kantr | 35.8 | 9.6 | 5.3 | 1.8 | 0.070 | 0.467 |
| Tspyl2 | 124.9 | 11.4 | 4.6 | 2.5 | 0.013 | 0.298 |
| Tceanc | 27.9 | 9.2 | 5.6 | 1.6 | 0.102 | 0.508 |
ここでpvalueはWald検定のp値であり、padjは多重比較の誤りを考慮して補正されたp値です。
転写因子推定
さきほどと同じくCollectTRIを用います。
univariate linear model(ULM)による推論を行います。
ULMでは1つの目的変数を予測するための線形モデルです。このモデルの特徴は変数間の相関関係も考慮にいれることです。転写因子推定では目的変数が一つなのでこのモデルを使用します。
# Infer TF activities with ulm
tf_acts, tf_pvals = dc.run_ulm(mat=mat, net=collectri, verbose=True)
tf_acts
結果は以下のようになります。
上位の活性/不活性転写因子について取得したスコアをプロットします。
dc.plot_barplot(
acts=tf_acts,
contrast='treatment.vs.control',
top=25,
vertical=True,
figsize=(3, 6)
)
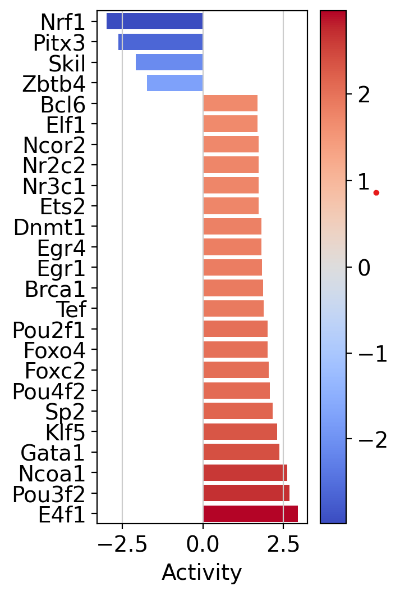
activityはp値をもとに算出した値だと思われます。
APT_IDマウスでは、E4f1, Pou3f2等が活性化され、Nrf1, Pitx3等は不活性化されるようです。
これらの遺伝子を調べていきます。
E4f1
Among its related pathways are p53 pathway.
May function as a transcriptional repressor. May also function as a ubiquitin ligase mediating ubiquitination of chromatin-associated TP53. Functions in cell survival and proliferation through control of the cell cycle.
今回あまり関係がないように思われる。
Pou3f2
Among its related pathways are Nervous system development and Neuroscience.
これも特にヒットせず。
Nrf1
The protein has also been associated with the regulation of neurite outgrowth.
神経突起延長に関連する。これが阻害されて神経突起延長に支障がでるというのは理解できる。
Pitx3
Transcriptional regulator which is important for the differentiation and maintenance of meso-diencephalic dopaminergic (mdDA) neurons during development. In addition to its importance during development, it also has roles in the long-term survival and maintenance of the mdDA neurons. Activates NR4A2/NURR1-mediated transcription of genes such as SLC6A3, SLC18A2, TH and DRD2 which are essential for development of mdDA neurons.
ドーパミン作動性ニューロンの分化と維持に重要。アルツハイマー病ではドーパミンが減少していることが報告されているため、これが抑制されているのは理解できる。
まとめ:不活性化されている転写因子は、阻害されるとアルツハイマーにつながりうることが分かった。
次に転写因子のネットワーク をプロットし、アクティビティとターゲット遺伝子発現ごとにノードを色分けします。
dc.plot_network(
net=collectri,
obs=mat,
act=tf_acts,
n_sources=['Pou3f2', 'E4f1', 'Nrf1', 'Eno1'],
n_targets=15,
node_size=50,
figsize=(7, 7),
c_pos_w='darkgreen',
c_neg_w='darkred',
vcenter=True
)
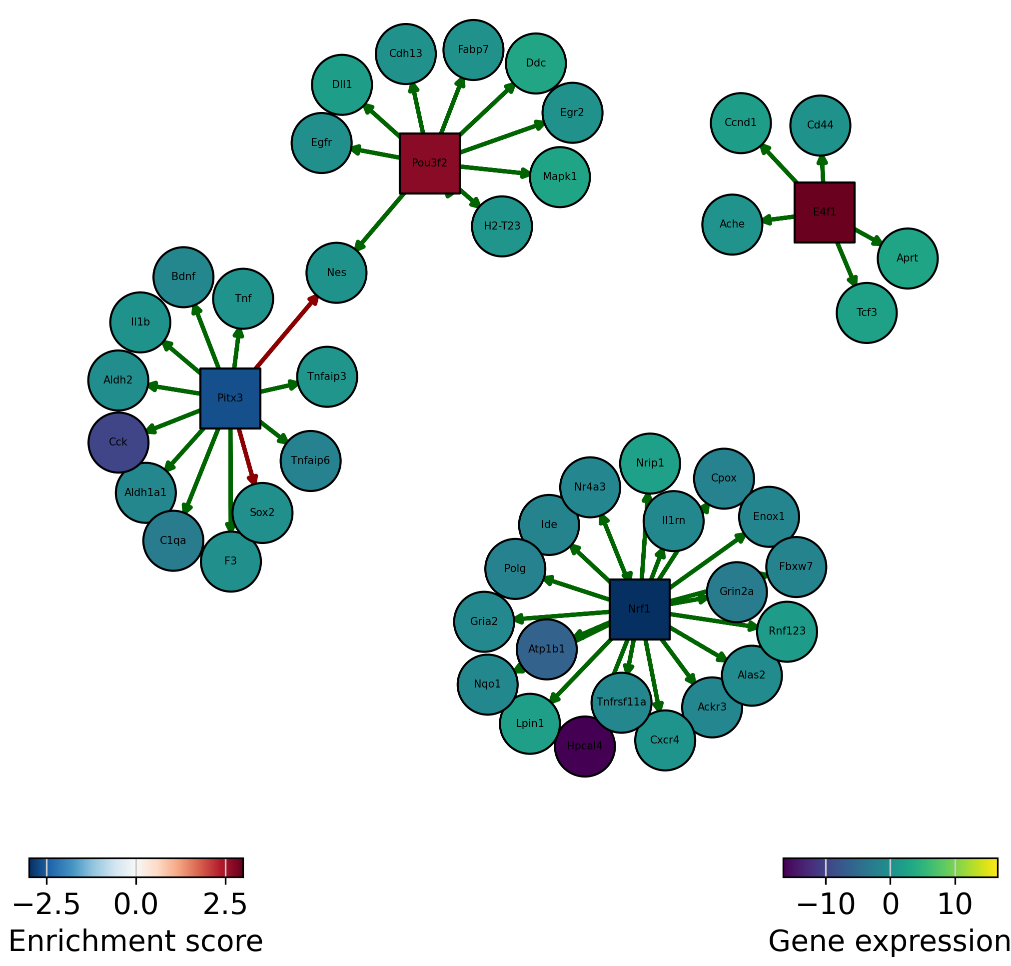
中心にある正方形のものが転写因子、外側にある丸いものが遺伝子を表しています。赤い矢印は抑制することを、緑は活性化することを表しています。
また、転写因子の発現度合いを表すのがEnrichment scoreで遺伝子発現を表すのがGene expressionです。
この図では不活性転写因子であるPitx3がNesという発現量は平均的な遺伝子を負に制御し、活性化転写因子であるPou3fwがNesを正に制御していることがわかります。
volcano plotを示します。

x軸は0を中心にして、右側が発現量が増加した遺伝子、左側が発現量が減少した遺伝子。y軸は上に行くほど統計的有意性が高い遺伝子を表す。
グループ間での発現の変動比が大きく、発現の変動の統計的有意性が高い遺伝子は、図の右上や左上に位置することになる。
Hpca4がどのような役割を果たす遺伝子なのかはよくわからない。
パスウェイ解析
同様にPROGENyを用います。
データベースをダウンロードして解析します。
multivariate linear model(MLM)を使用します。
MLMは複数の目的変数を同時に予測するための線形モデルで、特徴は複数の目的変数を扱うことです。また、変数間の相関関係も考慮にいれます。
パスウェイ解析では複数の遺伝子発現量を同時に目的変数として扱うためにこのモデルを使用します。
# Infer pathway activities with mlm
pathway_acts, pathway_pvals = dc.run_mlm(mat=mat, net=progeny, verbose=True)
pathway_acts
この後図示すると以下のようになります。

JAK-STATやEGFRが活性化している。p53やEstrogenは不活性化している。
EGFR : 哺乳類細胞の成長、生存、遊走、アポトーシス、増殖、分化を調節します。
Estrogen: 女性の生殖器の成長と発達を促進します。
JAK-STAT : 免疫、細胞分裂、細胞死、腫瘍形成に関与します。
p53 : 細胞周期、アポトーシス、DNA修復、腫瘍抑制を制御します。
これより活性化しているJAK-STATやEGFRは免疫亢進(免疫寛容)に働き、不活性化している
p53やEstrogenは癌化促進などに働くことがわかる。(Estrogenは謎)
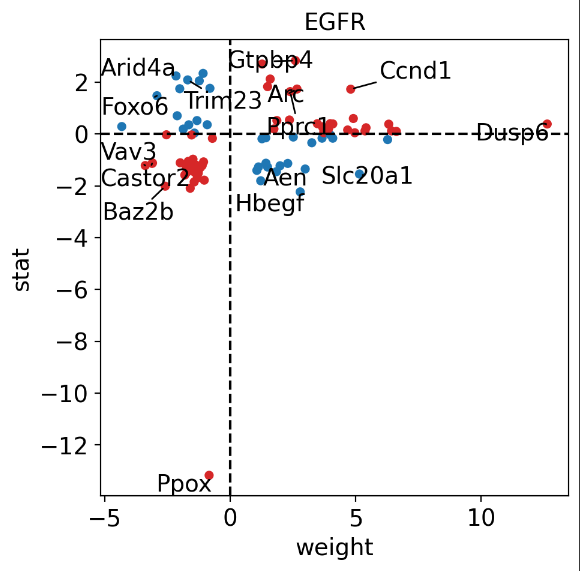
横軸のweightは、パスウェイ(ここではEGFR)における各遺伝子の重みを表しています。
statはワルド統計量(2乗していないもの)です。ワルド統計量の5%値は3.84であり、今回のstatではルートすればよいので1.95です。ここらへんに5%のラインがあると考えてください。
生物学的プロセス推定
同様にMSigDBを用います。データベースをダウンロードしてORAで解析します。
# Infer enrichment with ora using significant deg
top_genes = results_df[results_df['padj'] < 0.01]
# Run ora
enr_pvals = dc.get_ora_df(
df=top_genes,
net=Mus_musculus_msigdb,
source='geneset',
target='genesymbol'
)
enr_pvals.head()
この後図示すると以下のようになります。
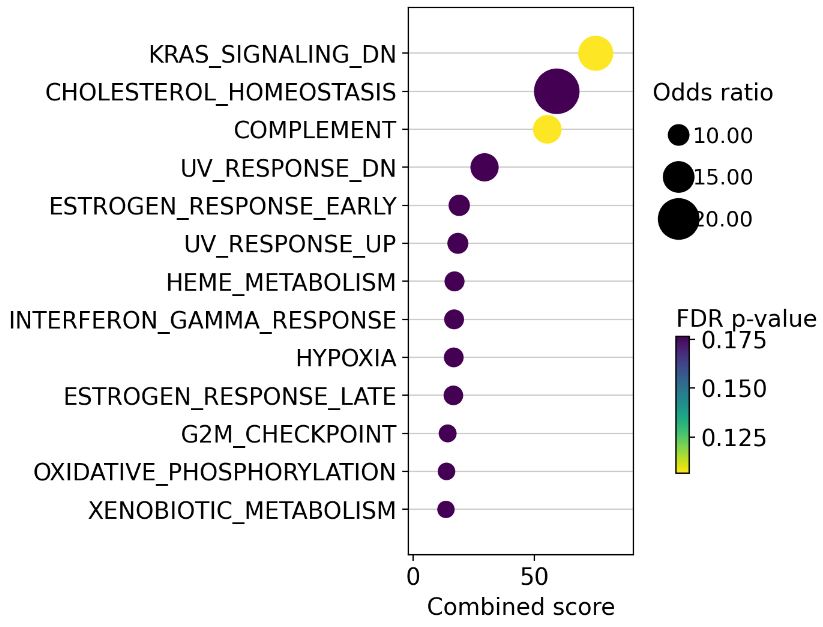
KRAS_SIGNALING_DNとCOMPLEMENTが強化されていると思われる。
KRAS_SIGNALING_DN
KRAS is a proto-oncogenic GTPase
KRAS_SIGNALING_DNはKRAS活性化を抑制するため癌化を抑制している
COMPLEMENT
Genes encoding components of the complement system, which is part of the innate immune system.
よって免疫系が活性化していると考えられる
まとめ
目標は以下であった。
APT_IDでは免疫寛容がおこっている。ミクログリア、CD73(NT5E)、アデノシンの減少、Treg細胞亢進、TMEM119| CD11bとCD45| Iba1| CX3CR1| F4/80 | CD68 | CD40減少を示したい
得られた結果は以下。
- EGFRに注目するとWD_CDでKupper cellとMacrophagesという血管透過性を必要とする細胞で発現しているのに対し、APT_IDではBasophils(好塩基球),Kupper cell, Monocytesで発現しており何らかの免疫反応が亢進していることがうかがえる
- JAK-STATはAPT_IDのMonocytesでよく発現している。JAK-STATは免疫を強くする作用があるため、APT_IDのMonocytesにおいて免疫反応が亢進されていることがわかる
- APT_IDで不活性化されている転写因子の中に、阻害されるとアルツハイマーにつながるものがある
- APT_IDで活性化している転写因子の中に免疫亢進(免疫寛容)に働くものがあり、不活性化している転写因子の中に癌化抑制(結果的に癌化促進)などに働くものがある
- APT_IDで亢進している遺伝子には、癌化を抑制するもの、免疫を活性化させるものがある
これらの結果から免疫寛容が起こっていそうなことがわかる。しかしながらミクログリア、CD73(NT5E)、アデノシンの減少、Treg細胞亢進、TMEM119| CD11bとCD45| Iba1| CX3CR1| F4/80 | CD68 | CD40減少という分子機構のふるまいについてはわからなかった。
しかし、これを書いているあたりでGSE219130の元論文が探してもみつからないことが発覚した。もしかしたら論文としてだせなかったものの可能性も出てきた。そういう意味では、直接的に示せなかったのはある意味正しかったかもしれない。
最後に
反省点は主に3点あります。1つ目はアノテーションを自分の手で正確に行うことができなかったこと。2つ目は適切なフィルタリングができていなかった可能性が高いこと。3つめは元論文を最初に確認しなかったためにそもそもうまくいかないことをやってしまっていた可能性があることです。論文を選ぶ際は適切なジャーナルから引っ張ってこないといけないと痛感しました。
しかしsingle cellの解析や個体ごとの解析を通じて、遺伝子解析がどういったものであるのか見えてきました。また、pythonにも多少親しむことができました。
次は正しい論文を用いて同じことをしてみたいです。
(この記事は研究室インターンで取り組みました:https://kojima-r.github.io/kojima/)