こんにちは。仕事でシングルセル解析を行う予定なので、勉強したことをこれからまとめてきます。
シングルセル解析とは
検体中に含まれる細胞や細菌等の微生物をシングルセル、つまり細胞ごとに分離してゲノム情報や機能を解析し、データを得る手法のこと。

引用:dynacom 29. シングルセル解析: COVID-19患者と健常者の比較解析 Fig.1 解析ステップごとの解析内容及び主な解析ツール
手順は以下。
- 初めに酵素処理やフローサイトメトリー(FACS)などの技術を用いて細胞の単離を行い、逆転写酵素を使ってmRNAからcDNAを合成する。その後、次世代シークエンス(NGS)技術で塩基配列を解読する。→ これにより、どの遺伝子情報がどの細胞由来なのかを区別できる
- 一次解析として、シークエンスデータのマッピング、発現カウントを行う
- 二次解析では、遺伝子発現データに基づいて細胞のクラスター分析を行う。またマーカー遺伝子(特定の細胞種を識別するための遺伝子)の発現量に基づき、各クラスターの細胞種を同定する(=アノテーション)
- さらに細胞種の構成比や遺伝子発現量を確認することで、細胞集団内・間の差異を評価する
一次解析では10x Genomics社がサポートする「Cell Ranger」、二次解析では統計解析ソフトRのパッケージ「Seurat」が広く使用されています。
高次解析では、二次解析で得られた結果を用いてGO解析やパスウェイ解析を行い、より詳細な生物学的意義を調べていきます。
今回は、上記のシングルセル解析の中でも、アノテーションを扱います。
アノテーションとは
シングルセル解析では、サンプルの細胞種が不明な状態でデータが得られるため、データから細胞種を推定することが必要となります。
このように得られた情報から細胞種を推定し、各サンプルに情報を割り当てることをアノテーションと言います。
では、さっそく実装してみましょう!
使用言語・ツール・データ
- 使用言語:Python
- ライブラリ:Scanpy, CellTypist, decoupler
-
データ:健康の人の骨髄単核細胞
- 以下のscanpyのチュートリアルで扱っているデータと同じもの
https://scverse-tutorials.readthedocs.io/en/latest/notebooks/basic-scrna-tutorial.html
- 以下のscanpyのチュートリアルで扱っているデータと同じもの
アノテーションツールの種類
アノテーションの方法には様々な手法がありますが、今回は以下の3つを順番に試していきたいと思います。
- 手動アノテーション(Scanpyチュートリアルに基づく)
- decoupler
- CellTypist(機械学習を用いた自動アノテーション)
前処理
ここから実際にアノテーションを行うまでの手順がとても長いので、どのようなことを行うかを最初に示します。

ライブラリのインポート
import scanpy as sc
import anndata as ad
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import scipy.io
import celltypist
from celltypist import models
import decoupler as dc
準備
DOIリポジトリからデータファイルを取得するための準備です。
# リモートからデータファイルを取得し、ローカルにキャッシュする
EXAMPLE_DATA = pooch.create(
path=pooch.os_cache("scverse_tutorials"), # キャッシュフォルダを指定
base_url="doi:10.6084/m9.figshare.22716739.v1/", # BMNCの生データ(10x Genomics フォーマットの HDF5) が保存されているDOIリポジトリ
)
EXAMPLE_DATA.load_registry_from_doi()
データのダウンロード
デフォルトでは pooch.os_cache("プロジェクト名") にキャッシュが保存されます。
- Windows → C:\Users\ユーザー名\AppData\Local\myproject\
- Linux/macOS → ~/.cache/myproject/
- キャッシュフォルダには、取得したデータファイルと pooch の管理情報が含まれる
samples = {
"s1d1": "s1d1_filtered_feature_bc_matrix.h5", # Day 1(1日目)のBMNCデータ
"s1d3": "s1d3_filtered_feature_bc_matrix.h5", # Day 3(3日目)のBMNCデータ
}
adatas = {}
# pooch を使ってデータをダウンロードし、ローカルにキャッシュ(ダウンロード済みなら キャッシュされたファイルをそのまま利用)
for sample_id, filename in samples.items():
path = EXAMPLE_DATA.fetch(filename) # fetchはリモートにあるデータをダウンロードし、ローカルのキャッシュから利用するためのメソッド
sample_adata = sc.read_10x_h5(path) # 10x Genomics フォーマットを読み込む
sample_adata.var_names_make_unique() # 遺伝子名の重複がある場合は区別
adatas[sample_id] = sample_adata # 読み込んだデータを辞書に保存
# AnnDataを統合
adata = ad.concat(adatas, label="sample")
adata.var_names_make_unique() # 変数名(遺伝子名)をユニークにする
adata.obs_names_make_unique() # 観測データ(細胞ID)をユニークにする
print(adata.obs["sample"].value_counts())
データの概要
print("--adataの結果--\n", adata)
print("--adata.varの結果--\n", adata.var)
print("--adata.obsの結果--\n", adata.obs)
print("--adata.Xの結果--\n", adata.X)
上記により、このような情報が分かります。
--adataの結果--
AnnData object with n_obs × n_vars = 17125 × 36601
obs: 'sample'
--adata.varの結果--
Empty DataFrame
Columns: []
Index: [MIR1302-2HG, FAM138A, OR4F5, AL627309.1, AL627309.3, AL627309.2, AL627309.5, AL627309.4, AP006222.2, AL732372.1, OR4F29, AC114498.1, OR4F16, ...]
[36601 rows x 0 columns]
--adata.obsの結果--
sample
AAACCCAAGGATGGCT-1 s1d1
AAACCCAAGGCCTAGA-1 s1d1
AAACCCAAGTGAGTGC-1 s1d1
AAACCCACAAGAGGCT-1 s1d1
AAACCCACATCGTGGC-1 s1d1
... ...
TTTGTTGAGAGTCTGG-1 s1d3
TTTGTTGCAGACAATA-1 s1d3
TTTGTTGCATGTTACG-1 s1d3
TTTGTTGGTAGTCACT-1 s1d3
TTTGTTGTCGCGCTGA-1 s1d3
[17125 rows x 1 columns]
--adata.Xの結果--
<Compressed Sparse Row sparse matrix of dtype 'float32'
with 26550469 stored elements and shape (17125, 36601)>
Coords Values
(0, 43) 3.0
(0, 59) 1.0
(0, 60) 1.0
(0, 62) 1.0
(0, 72) 1.0
: :
(17124, 36567) 2.0
(17124, 36568) 24.0
(17124, 36569) 6.0
(17124, 36570) 1.0
(17124, 36571) 48.0
AnnDataとは?
ここで聞きなじみのない名前が出てきましたので補足します。
Scanpyでは、scRNA-seqデータをAnnDataオブジェクトとして扱います。
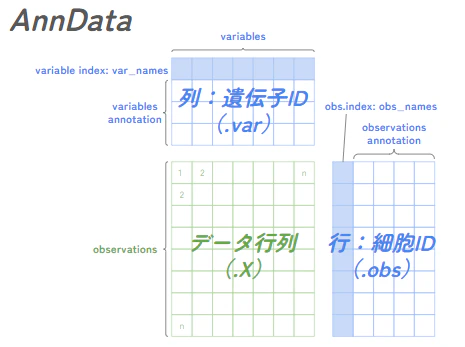
データ行列を中心に、ver_namesをキーに遺伝子の情報が、obs_namesをキーに細胞の情報がくっついているイメージです。
-
AnnData.var
- 遺伝子の情報(遺伝子ID, 遺伝子名)
- 遺伝子IDはEnsemblというサイトで定義された識別番号らしいです
https://asia.ensembl.org/index.html
-
AnnData.obs
- 細胞の情報(細胞バーコード)
-
AnnData.X
- 遺伝子発現データの行列(遺伝子 × 細胞)
なお、上記はあくまで基本構造であり、ここから各細胞や遺伝子に関する様々なメタデータを obs や var に保存できます。
そのほかにも、uns や obsm もあり、uns には解析結果に関する追加情報が辞書型で格納され、obsm には細胞ごとの低次元データ(例:PCA, UMMAPの座標情報)が格納されます。
クオリティコントロ-ル
シングルセル RNA シーケンシング(scRNA-seq)データには、細胞の品質のばらつき や 技術的なノイズ が含まれているため、それを除去して解析の精度を上げるのが クオリティコントロール(QC)の目的です。
一般的には以下の3つをチェックします。
-
nFeature_RNA(遺伝子数)
- 各細胞で 検出された遺伝子の数(特徴量の数)
- 極端に少ない(例: < 200)場合は、死んでいる細胞や RNA の分解の可能性がある
- 極端に多い場合は、ダブルレット(二重細胞) の可能性がある
-
nCount_RNA(発現量の合計)
- 各細胞における 全遺伝子の発現量の合計(合計カウント数)
- 極端に低いと デブリ(細胞が壊れている) の可能性がある
- 極端に高いと、ダブルレット(二重細胞) の可能性がある
-
percent_mt(ミトコンドリア遺伝子比率)
- 各細胞におけるミトコンドリア遺伝子の発現割合(%)
- 通常 5〜10% 以下 に抑えるのが一般的
- 高すぎる(>10-15%)場合は、死にかけの細胞 の可能性がある
特定の遺伝子IDから始まるもものか否かのbool値をadata.varに追加します。
# ミトコンドリア遺伝子, "MT-" は人間, "Mt-" はねずみ
adata.var["mt"] = adata.var_names.str.startswith("MT-")
# リボソーム遺伝子
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
# ヘモグロビン遺伝子
adata.var["hb"] = adata.var_names.str.contains("^HB[^(P)]")
追加した結果は以下のようになります。
print(adata.var)
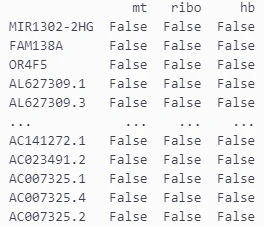
今まではインデックスの列に遺伝子IDがあるだけでしたが、それぞれのカラムが追加されていることが分かります。
次に、先ほどの3つの遺伝子集団の発現割合を調べてみます。
# 特定の遺伝子集団をcalculate_qc_metrics()に渡して、その発現割合を計算する
sc.pp.calculate_qc_metrics(
adata,
qc_vars=["mt", "ribo", "hb"],
inplace=True, # 計算結果が adata.obs に直接追加される
log1p=True # 自然対数変換(log1p(x) = log(1 + x))
)
バイオリンプロット
それぞれを可視化してみます。※チュートリアルでは、さきほど挙げた3つ(遺伝子量、発現量の合計、ミトコンドリアの割合)しか可視化していません。
sc.pl.violin(
adata,
[
"total_counts", # 細胞あたりの合計カウント(adata.Xの各行ごとの合計カウントと一致)
"total_counts_mt",
"total_counts_ribo",
"total_counts_hb"
],
jitter=0.4,
multi_panel=True,
)

sc.pl.violin(
adata,
[
"pct_counts_mt",
"pct_counts_ribo",
"pct_counts_hb"
],
jitter=0.4,
multi_panel=True,
)
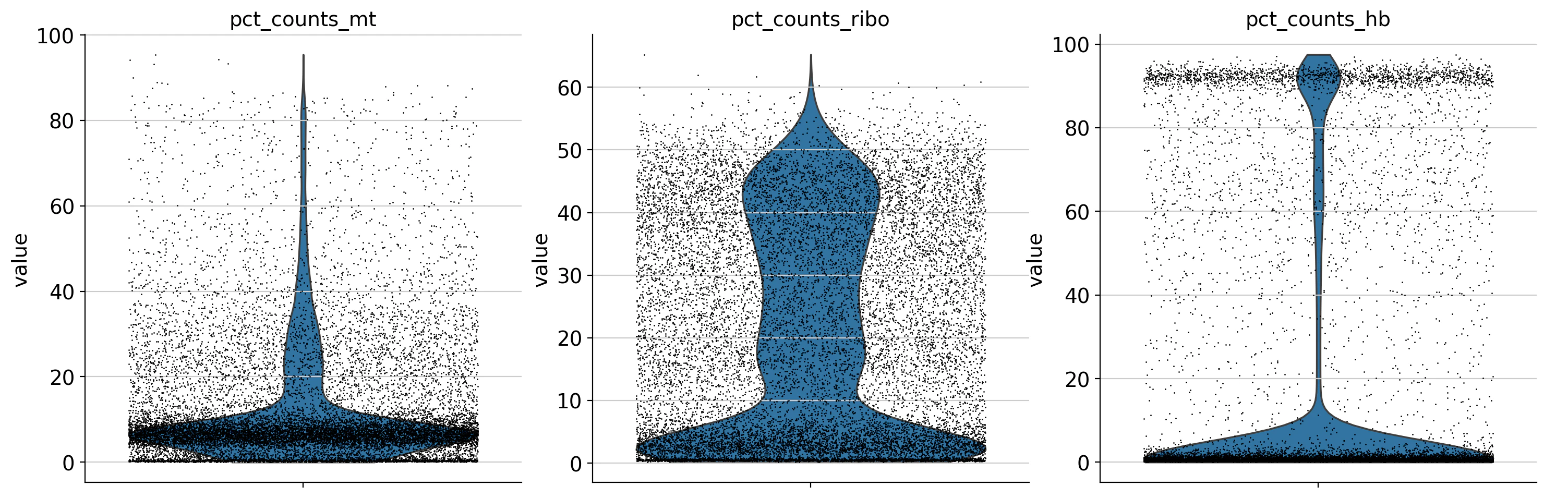
細胞や遺伝子に関する知識をもとに閾値を決めます。
散布図
total_counts(細胞のRNA発現総カウント)と n_genes_by_counts(発現遺伝子数)の関係を可視化してみます。また、pct_counts_mt(ミトコンドリア遺伝子の割合)の影響も見てみます。
sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt")
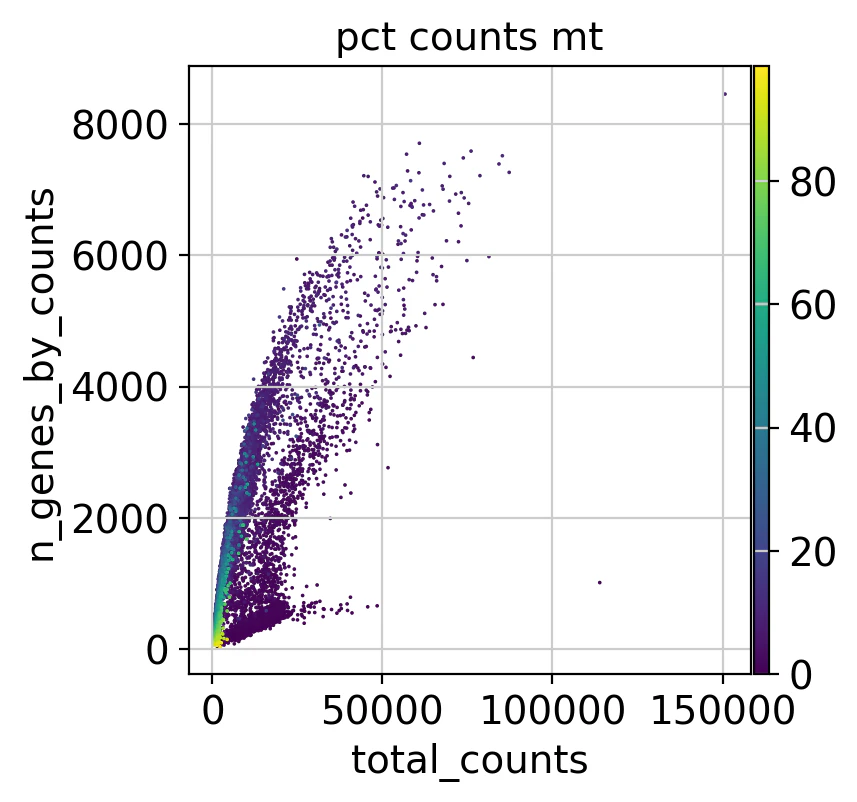
細胞あたりの総カウント数(total_counts)が増えると、発現する遺伝子数(n_genes_by_counts)も増える傾向にあることが分かります。つまり、より多くのRNAを持つ細胞はより多くの遺伝子が発現している可能性が高いです。
また、ミトコンドリア遺伝子の割合との関係を見ると、総カウント数が低い細胞ほどミトコンドリア遺伝子の割合が高いことが分かります。これらがQCフィルタリングの対象となりそうです。
QCフィルタリング
ここでは、チュートリアルに則り、まずおおざっぱにフィルタリングします。
sc.pp.filter_cells(adata, min_genes=100) # 発現する遺伝子が100個未満の細胞を除外する(死細胞の可能性)
sc.pp.filter_genes(adata, min_cells=3) # 発現している細胞数が3個未満の遺伝子を除外する(ノイズやエラーの可能性)
結果として、 n_obs × n_vars = 17041 × 23427となりましたので確かに減っています。
ダブレット検出
ダブレットとは、2つまたはそれ以上の細胞が偶然一緒にキャプチャされ、単一の細胞として処理される現象を指します。これは、異なる細胞タイプからの遺伝子発現パターンを混在させることになるので、解析を混乱させる可能性があります。
これらのダブレット検出によるフィルタリングについては、DoubletFinder、Scrublet、Soloなどのツールがあります。
今回は、Scurbletを使います。
sc.pp.scrublet(adata, batch_key="sample") # データセットに複数のサンプル(異なる実験・バッチ) が含まれている場合、それぞれのサンプルごとにダブレットを検出
原理
- 実際の細胞の遺伝子発現データをランダムに組み合わせて 疑似的なダブレットを作成
- 細胞ごとのダブレットスコアの計算
各細胞がダブレットである可能性を ダブレットスコア(0〜1の連続値) で表す(adata.obs["doublet_score"]に保存される) - 閾値を設定してダブレットを分類
スコアが高い細胞をダブレットとしてフラグ付け(adata.obs["predicted_doublet"] = True/False)。
※デフォルトでは Scrublet が自動で適切な閾値を決定 するが、手動で調整可能 - この結果、`dobrel_score`(各細胞がダブレットである確率(0〜1))と、
predicted_doublet(True(ダブレット)、False(シングルト))がadata.obsに追加される
実際にダブレットスコアがどのような分布になっているかも見てみましょう。
sc.pl.scrublet_score_distribution(adata)
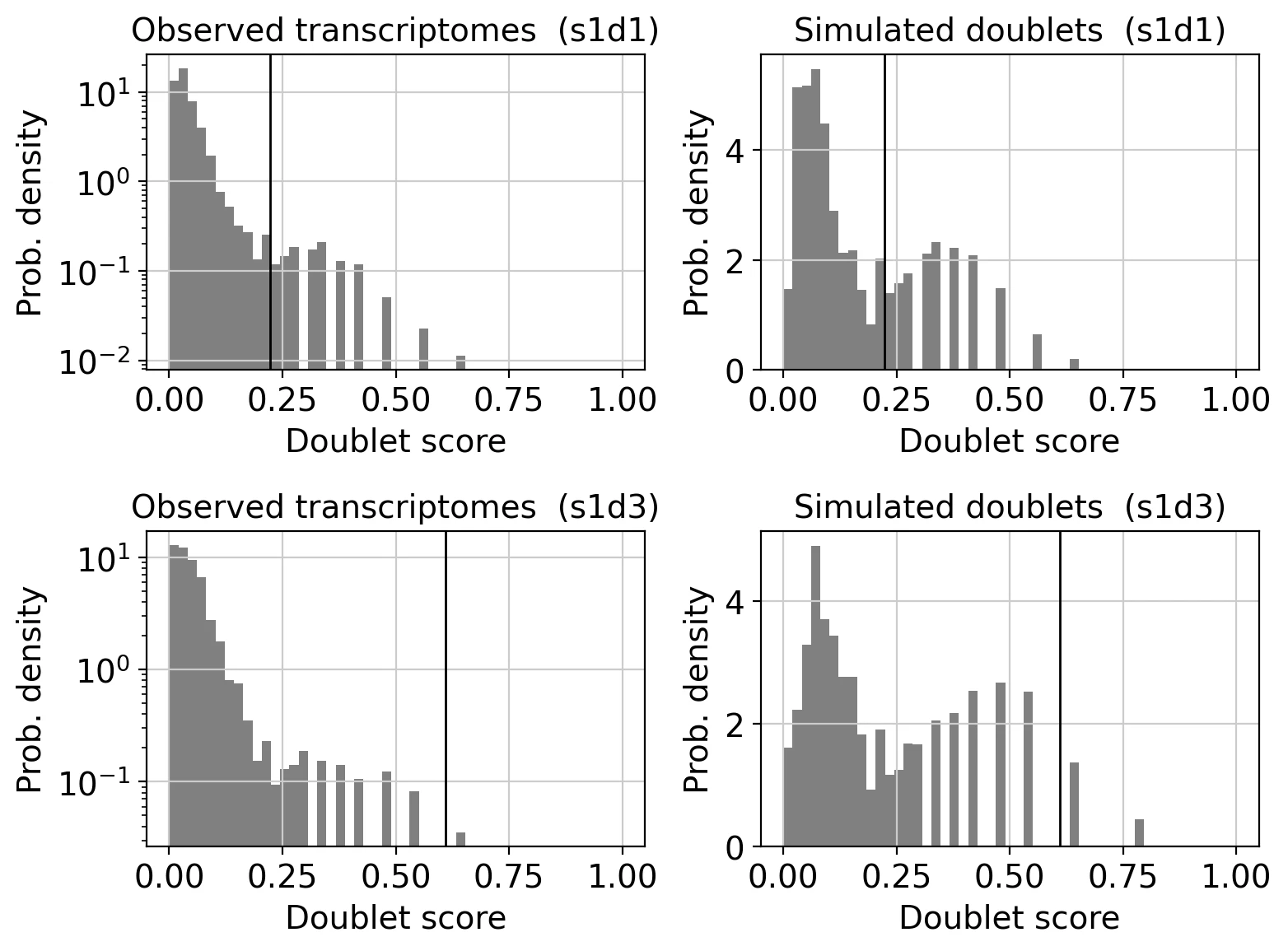
図の見方として、左は観測データになっており、実際のデータから計算したダブレットスコアの分布です。
右はシミュレーションで、ランダムに生成したダブレットスコアの分布になっています。
このようなダブレットは、細胞をフィルタリングして削除するか、後ほど行うクラスタリングをしてダブレットスコアが高いクラスターを削除するといった方法があります。
チュートリアルでは、クラスタリングの最後にQCを再評価しています。
※ 後からフィルタリングを行う場合は、元のカウントを保持しておきます。
# カウントを保存(adata.X は 正規化・対数変換 などの処理で上書きされるため、生データ(UMI カウント)を保存)
adata.layers["counts"] = adata.X.copy()
正規化と対数変換
合計カウントの正規化および対数変換をしていきます。
# 遺伝子発現カウントを正規化
sc.pp.normalize_total(adata) # デフォルトでは、細胞ごとのカウントの合計が 1e4(= 10,000)になるように正規化
# 対数変換
sc.pp.log1p(adata)
-
正規化
細胞ごとのRNAの量が異なると直接比較が難しい → 全細胞のそうカウントを統一することで細胞間の違いを補正しやすくする -
対数変換
一部の遺伝子の発現が極端に高くなる → 対数変換を適用することで、極端に高い発現値の影響を抑える
高変動遺伝子を選択
データは数千~数万の遺伝子を含みますが、すべての遺伝子が同じくらい重要とは限りません。変動が大きい遺伝子を選択してそれに基づいて解析を行いましょう。
# 可変性の高い遺伝子に注釈をつける(Seurat(デフォルト),Cell Ranger,Seurat v3)
sc.pp.highly_variable_genes(adata, n_top_genes=2000, batch_key="sample")
異なるサンプルの場合は、batch_keyを設定し、バッチ間で共通して変動の大きい遺伝子を統合します。
これを実行すると、adata.varに新しくhighly_variableが追加されます。
では、その効果を可視化してみましょう。
sc.pl.highly_variable_genes(adata)

このままでは分かりづらいので注釈をつけました。

通常、発現量が高い遺伝子ほど分散が大きくなりますが、ただ発現量が高いだけでなく、そのばらつきが大きい遺伝子を選ぶことが重要です(細胞ごとの違いを捉えやすいため)。
よって、正規化した分散を使って高変動遺伝子を選択します。
Scanpyを使った手動アノテーション
今までは前処理を行ってきました。次から、手動およびdecouplerと、celltypistで処理の順番が異なってきます。
celltypistではスケーリング・次元削減前のデータを用いるのでここで一度保存しておきます。
adata.layers['before_DimRed'] = adata.X.copy()
次元削減
では、次元削減を行います。
sc.tl.pca(adata)
adata.Xの遺伝子発現データに対して、PCAを実行することで、データを低次元に変換し計算効率を向上させます。
この結果は、adata.obsm["X_pca"]に保存されます。
それでは、50次元に削減したデータの寄与率を見てみましょう。
sc.pl.pca_variance_ratio(adata, n_pcs=50, log=True) # 50PCsに変換
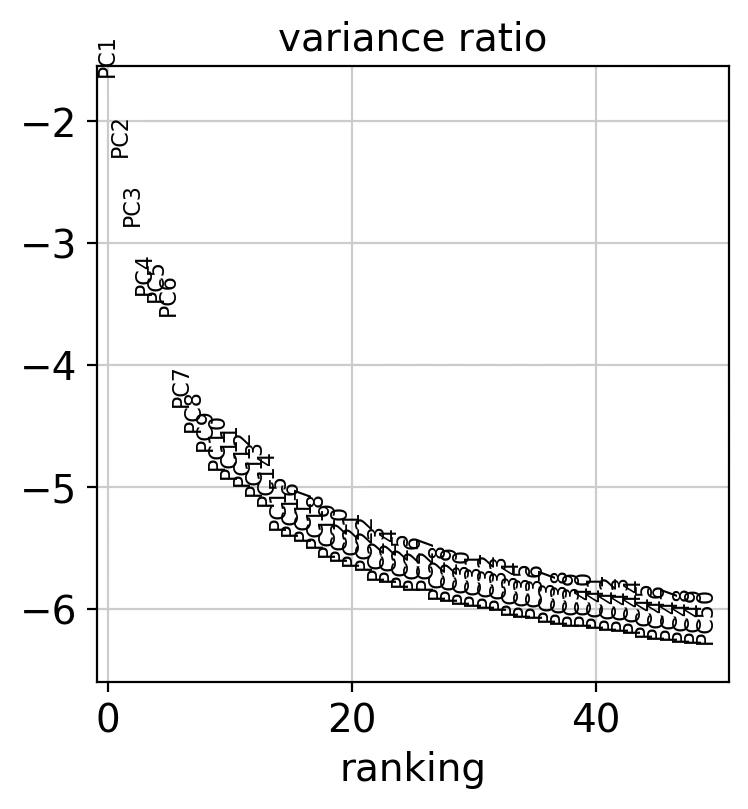
この可視化によって各主成分がデータの変動をどれくらい説明しているかを可視化しています。一般的には、エルボーポイントを見て適切な次元の数を選びます。
次にPCA空間における細胞の分布を可視化してみます。
sc.pl.pca(
adata,
color=["sample", "sample", "pct_counts_mt", "pct_counts_mt"],
dimensions=[(0, 1), (2, 3), (0, 1), (2, 3)],
ncols=2,
size=2,
)
バッチ効果(上段)やQC指標(下段)がデータの変動に影響していないかを確認するものです。
色が明確に分離されていたら、主成分の変動に影響を与えているということになります。
見たところ大丈夫なのかな?(チュートリアルはそのまま突き進む)
近傍グラフ
各細胞の最近傍(nearest neighbors)を探索し、グラフ構造を作ることで、類似度を数値的に表現します。
sc.pp.neighbors(adata)
PCAで次元を削減したデータ(adata.obsm["X_pca"])を使用し、各細胞に対して、最近傍の細胞(nearest neighbors)を見つけ、近傍グラフ(k-NN グラフ)を作成し、adata.obsp["distances"](距離行列) に保存します。
次元削減
この結果をUMAPを用いて二次元に削減してから可視化してみます。
なお、UMAPは局所的な構造を保持するための手法で計算不可が大きいので、最初にPCAで50次元に削減してからUMAPを適用するという順序を取っています。
sc.tl.umap(adata)
sc.pl.umap(
adata,
color="sample",
# 点のサイズを小さくしてオーバーラップを防ぐ
size=2,
)
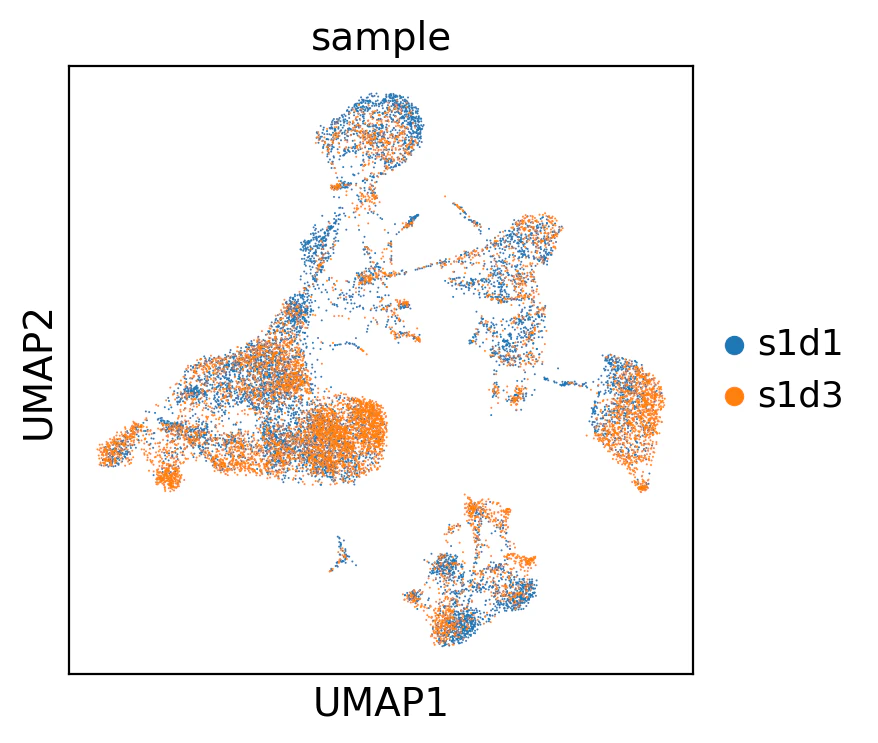
この可視化により、バッチごとに異なるクラスタが生じていないことが分かります。つまり、生物学的な差のほうがバッチ間の違いより大きいということです。
クラスタリング
では、最後にクラスタリングをして細胞のアノテーションを手動で行っていきます。
sc.tl.leiden(adata, flavor="igraph", n_iterations=2)
細胞が適切なクラスタに割り当てられたら、それをadata.obs['leiden']に保存します。
QCの再評価
クラスタリングやUMAPの結果を確認しながら、追加のQCを行うかどうかを判断します。
sc.pl.umap(
adata,
color=["leiden", "predicted_doublet", "doublet_score"],
wspace=0.5,
size=3,
)
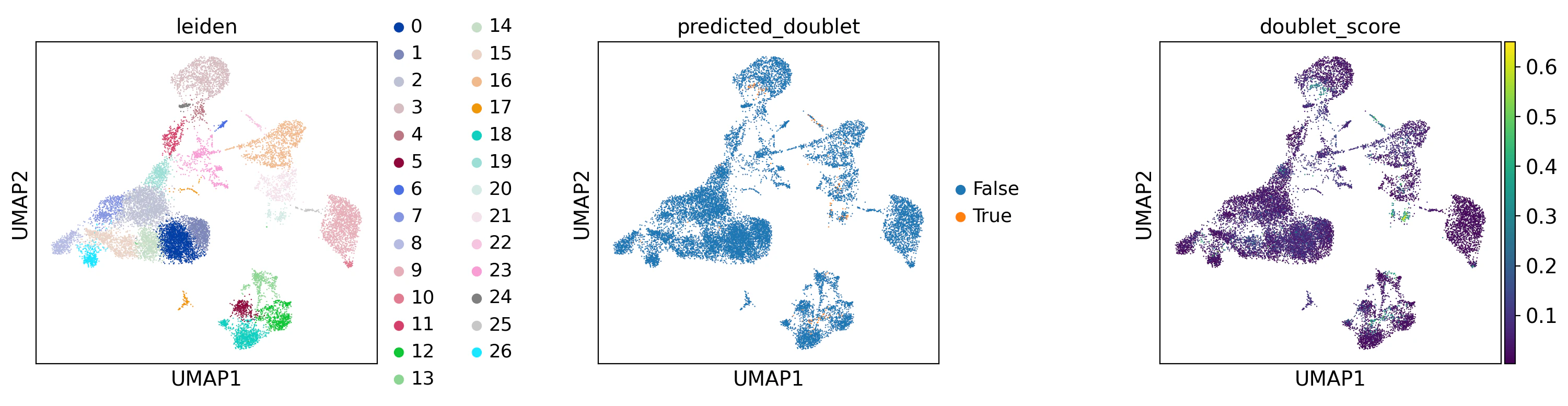
sc.pl.umap(
adata,
color=["leiden", "log1p_total_counts", "pct_counts_mt", "log1p_n_genes_by_counts"],
wspace=0.5,
ncols=2,
)
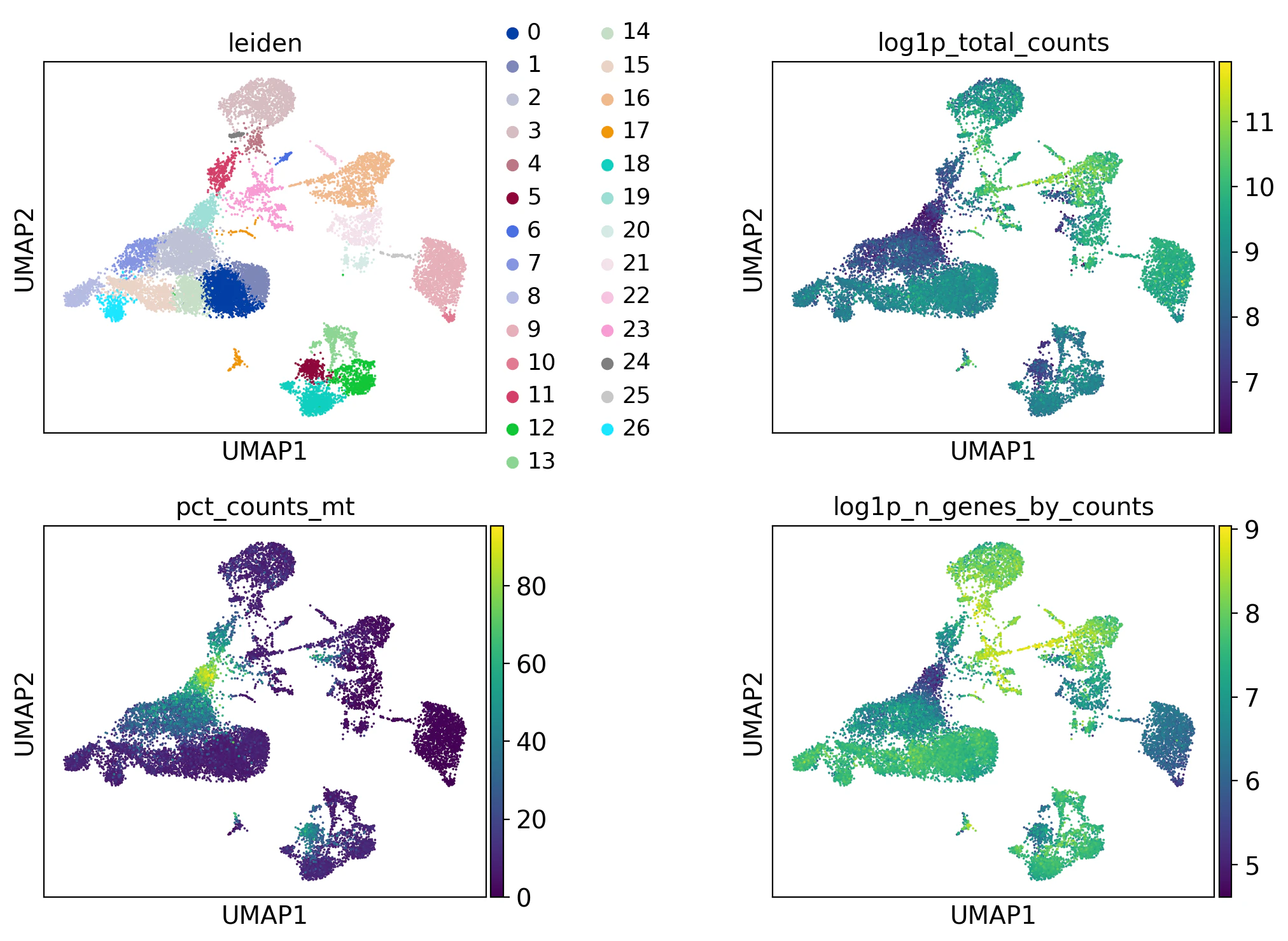
チュートリアルでは、この後深堀をしていなかったのですが、見た限り、特にミトコンドリアの割合がクラスターで明確に色が変わっており怪しいのでは..と個人的に思ったので数値でも見てみます。
まず、ダブレット割合を数値で確認します。
# クラスタごとのダブレット割合を確認
adata.obs.groupby("leiden")["predicted_doublet"].mean()
出力結果
leiden
0 0.000488
1 0.000000
2 0.000987
3 0.030303
4 0.000000
5 0.009554
6 0.000000
7 0.000000
8 0.006508
9 0.000000
10 0.013699
11 0.003390
12 0.043609
13 0.007752
14 0.004860
15 0.001045
16 0.011407
17 0.000000
18 0.025330
19 0.009804
20 0.324324
21 0.027140
22 0.377049
23 0.001949
24 0.000000
25 0.000000
26 0.000000
Name: predicted_doublet, dtype: float64
クラスタ22のダブレットスコアは37.7%でした。
割合にすると思ったより高くないですが、クラスタごとよりも以下のように個々に抜いていったほうがよいかもです、今回は行いませんが、個々に削除する場合はこちらのコードです。
adata = adata[adata.obs["predicted_doublet"] == False].copy()
次にミトコンドリアの割合を数値で見てみます。
# クラスタごとのミトコンドリア比率を確認
adata.obs.groupby("leiden")["pct_counts_mt"].mean()
出力結果
leiden
0 6.412249
1 7.356223
2 27.215322
3 8.199389
4 7.279397
5 33.692799
6 5.451702
7 24.767744
8 8.602679
9 0.419712
10 0.147228
11 40.633034
12 9.358269
13 7.485861
14 7.166812
15 8.876131
16 6.336304
17 13.759064
18 9.615798
19 67.817841
20 3.055290
21 2.767341
22 4.869937
23 11.826223
24 8.633160
25 1.132280
26 7.917830
クラスタ19はミトコンドリアの発現割合が67.8%と高く、遺伝子の発現量も低いことから怪しいです。
なお、今回はチュートリアルに則って削除しません。
アノテーションまでの手順をまとめると以下のとおりです。

手動による細胞タイプのアノテーション
アノテーションのおおまかな手順は以下のとおりです。チュートリアルではすべての細胞タイプのラベル付けをしておりません。
- Leidenクラスタリングを複数の解像度で実行
- 各クラスタごとに発現している遺伝子を調べる
- 細胞タイプごとのマーカー遺伝子と照らし合わせる
- 適切な細胞タイプをラベル付けする
解像度の調整
引数resolutionで解像度を調整します。
for res in [0.02, 0.5, 2.0]:
sc.tl.leiden(
adata, key_added=f"leiden_res_{res:4.2f}", resolution=res, flavor="igraph"
)
sc.pl.umap(
adata,
color=["leiden_res_0.02", "leiden_res_0.50", "leiden_res_2.00"],
legend_loc="on data",
)

ここではまだどの解像度が適切か結論づけません。
まず、主要な細胞タイプの遺伝子を設定します。
marker_genes = {
"CD14+ Mono": ["FCN1", "CD14"], # CD14+ Monocytes(単球)
"CD16+ Mono": ["TCF7L2", "FCGR3A", "LYN"], # CD16+ Monocytes(単球)
# 注意: DMXL2 は陰性マーカー
"cDC2": ["CST3", "COTL1", "LYZ", "DMXL2", "CLEC10A", "FCER1A"], # cDC2(古典型樹状細胞2)
"Erythroblast": ["MKI67", "HBA1", "HBB"], # Erythroblast(赤芽球)
# 注意: HBM と GYPA は陰性マーカー
"Proerythroblast": ["CDK6", "SYNGR1", "HBM", "GYPA"], # Proerythroblast(前赤芽球)
"NK": ["GNLY", "NKG7", "CD247", "FCER1G", "TYROBP", "KLRG1", "FCGR3A"], # NK(ナチュラルキラー細胞)
"ILC": ["ID2", "PLCG2", "GNLY", "SYNE1"], # ILC(自然リンパ球)
"Naive CD20+ B": ["MS4A1", "IL4R", "IGHD", "FCRL1", "IGHM"], # Naive CD20+ B cells(ナイーブB細胞)
# 注意: IGHD と IGHM は陰性マーカー
"B cells": [
"MS4A1",
"ITGB1",
"COL4A4",
"PRDM1",
"IRF4",
"PAX5",
"BCL11A",
"BLK",
"IGHD",
"IGHM",
], # B cells(B細胞)
"Plasma cells": ["MZB1", "HSP90B1", "FNDC3B", "PRDM1", "IGKC", "JCHAIN"], # Plasma cells(形質細胞)
# 注意: PAX5 は陰性マーカー
"Plasmablast": ["XBP1", "PRDM1", "PAX5"], # Plasmablast(形質芽球)
"CD4+ T": ["CD4", "IL7R", "TRBC2"], # CD4+ T(CD4+ T細胞)
"CD8+ T": ["CD8A", "CD8B", "GZMK", "GZMA", "CCL5", "GZMB", "GZMH", "GZMA"], # CD8+ T(CD8+ T細胞)
"T naive": ["LEF1", "CCR7", "TCF7"], # T naive(ナイーブT細胞)
"pDC": ["GZMB", "IL3RA", "COBLL1", "TCF4"], # pDC(形質性樹状細胞)
}
この遺伝子が含まれていたら、この細胞タイプの可能性が高いのではという目安です。
では、クラスタごとにどんな遺伝子が発現しているかをdotplotで可視化してみます。
# クラスタごとに発現している遺伝子を比較
sc.pl.dotplot(adata, marker_genes, groupby="leiden_res_0.02", standard_scale="var") # 遺伝子ごとに標準化を適用するオプション

- ドットの色 → 発現量の平均値(濃いほど高発現)
- ドットの大きさ → クラスタ内の発現細胞の割合
発現パターンが明確であるため、粗いクラスタリングではありますがラベリングします。
-
リンパ球
T細胞(CD4+, CD8+)、B細胞、NK細胞などを含む -
単球
マクロファージや樹状細胞へ分化する細胞 -
赤血球系
赤芽球(Erythroblast)や前赤芽球(Proerythroblast) から成熟赤血球へ分化 -
B細胞(B Cells)
ナイーブB細胞、メモリーB細胞、形質細胞(Plasma cells) に分類される
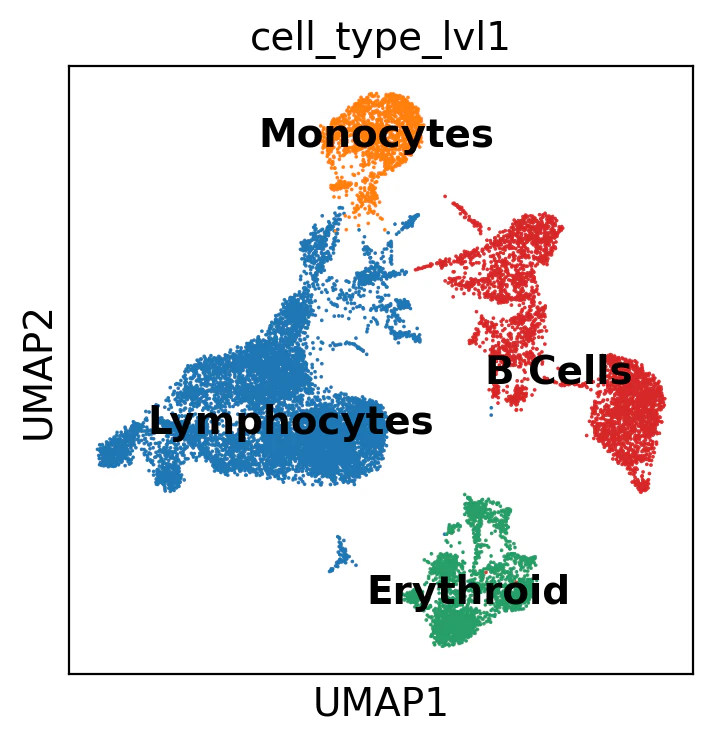
# Lidenクラスタを細胞タイプcell_type_lvl1に変換している
adata.obs["cell_type_lvl1"] = adata.obs["leiden_res_0.02"].map(
{
"0": "Lymphocytes", # リンパ球
"1": "Monocytes", # 単球
"2": "Erythroid", # 赤血球系
"3": "B Cells", # B B細胞
}
)
sc.pl.umap(adata, color="cell_type_lvl1", legend_loc="on data")
次に解像度0.5でもdotplotをしてみます。
sc.pl.dotplot(adata, marker_genes, groupby="leiden_res_0.50", standard_scale="var")

細胞タイプを区別するのに適したものとして、解像度0.5が適切とします。
また、今後行うdecouplerのためにデータを保存しておきます。
# decouplerはクラスタリング後のデータを用いるのでここで保存する
adata_clusterd = adata.copy()
差次的発言遺伝子をマーカーとして使用
あるクラスタで強く発現し、他のクラスタではほとんど発現しない遺伝子を特定する方法として、差次的発言遺伝子を見つけていきます。
各クラスタと残りのクラスタに対して、Wilcoxonランク和検定やt検定を実施します。
# 各クラスタごとに特異的に発現している遺伝子を統計的に計算
sc.tl.rank_genes_groups(adata, groupby="leiden_res_0.50", method="wilcoxon") # デフォルトではt検定
上位5位までの遺伝子をプロットします。
sc.pl.rank_genes_groups_dotplot(
adata, groupby="leiden_res_0.50", standard_scale="var", n_genes=5 # 各クラスタの上位5つ
)

※チュートリアルとはdotplotの結果(クラスタの順番?)が異なったので、この後のラベリングも同様に異なっていきます
ここでは、マーカー遺伝子の発現と差次的発言遺伝子が分かりやすいクラスタ8を取り上げたいと思います。
# 8のクラスタに特異的な遺伝子を取得
sc.get.rank_genes_groups_df(adata, group="8").head(5)
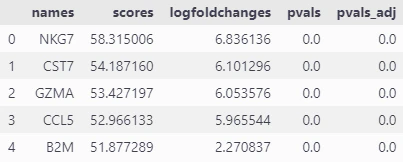
# クラスタ8の差次的発現遺伝子(DEGs)上位5つの遺伝子名を取得 し、dc_cluster_genes に格納
dc_cluster_genes = sc.get.rank_genes_groups_df(adata, group="8").head(5)["names"]
# UMAP でクラスタ8のDEGsの発現を可視化
sc.pl.umap(
adata,
color=[*dc_cluster_genes, "leiden_res_0.50"], # クラスタ8のDEGs+Leidenクラスタ
legend_loc="on data",
frameon=False,
ncols=3,
)

NKG7、CST7、GZMA、CCL5について、クラスタ8で発現割合が高い(黄緑)ことより、クラスタ8で特有にみられる遺伝子のようです。
以上より、クラスタ8はNK細胞もしくはCD8+T細胞が有力そうです。まだ決定づけるものが不足しているので、この後の別のアノテーション手法でも観察していきます。
さて、チュートリアルはここで記載が終わっていますが、せっかくならすべてのクラスタに手動アノテーションを試みたいと思います。
とは言え、一つ一つ調べていく時間はないので、今回はより細かいマーカー遺伝子のリストを取得してそれに当てはまった遺伝子を当てはめるようにしたいと思います。
今回は、PanglaoDBのデータセットを取得します。
import decoupler as dc # PanglaoDB のデータを取得するためのライブラリ
# PanglaoDB のデータセットを取得
markers = dc.get_resource(name="PanglaoDB", organism='human')
markers = markers[markers['human'].astype(bool) & markers['canonical_marker'].astype(bool) & (markers['human_sensitivity'].astype(float) > 0.5)]
# 不要な重複データを削除
markers = markers[~markers.duplicated(['cell_type', 'genesymbol'])]
# データの確認
print(markers.head())
# すべての細胞タイプと対応する遺伝子を取得
all_cell_types = markers["cell_type"].unique()
all_markers = markers.groupby("cell_type")["genesymbol"].apply(list)
all_markers
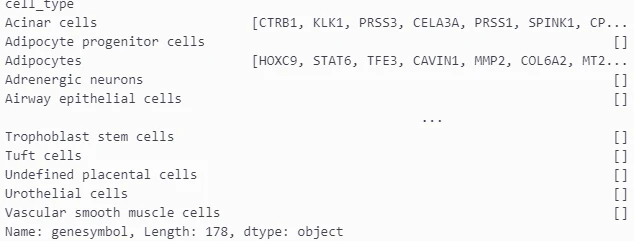
細胞タイプとそれに該当する遺伝子のリストを取得しました。
# 各クラスタのマーカー遺伝子リストを取得
top_genes_per_cluster = sc.get.rank_genes_groups_df(adata, group=None).groupby("group")["names"].apply(list)
top_genes_per_cluster

各クラスターに入っている遺伝子をリストで取得しました。
# クラスタごとに最も適合する細胞タイプを自動で割り当て
cell_type_map_all = {}
for cluster, genes in top_genes_per_cluster.items(): # 辞書オブジェクトのitems()メソッドを使い、各要素のキーと値に対してforループ処理
for cell_type, markers in all_markers.items():
if any(g in markers for g in genes[:5]):
cell_type_map_all[cluster] = cell_type
break
ここから、クラスタに含まれる上位5つの遺伝子が取得したマーカー遺伝子内にあればその細胞タイプを単純にcell_type_map_allに追加していくfor文をまわします。
adata.obs["cell_type_all"] = adata.obs["leiden_res_0.50"].map(cell_type_map_all)
adata.obs["cell_type_all"]
sc.pl.umap(
adata,
color="cell_type_all",
)
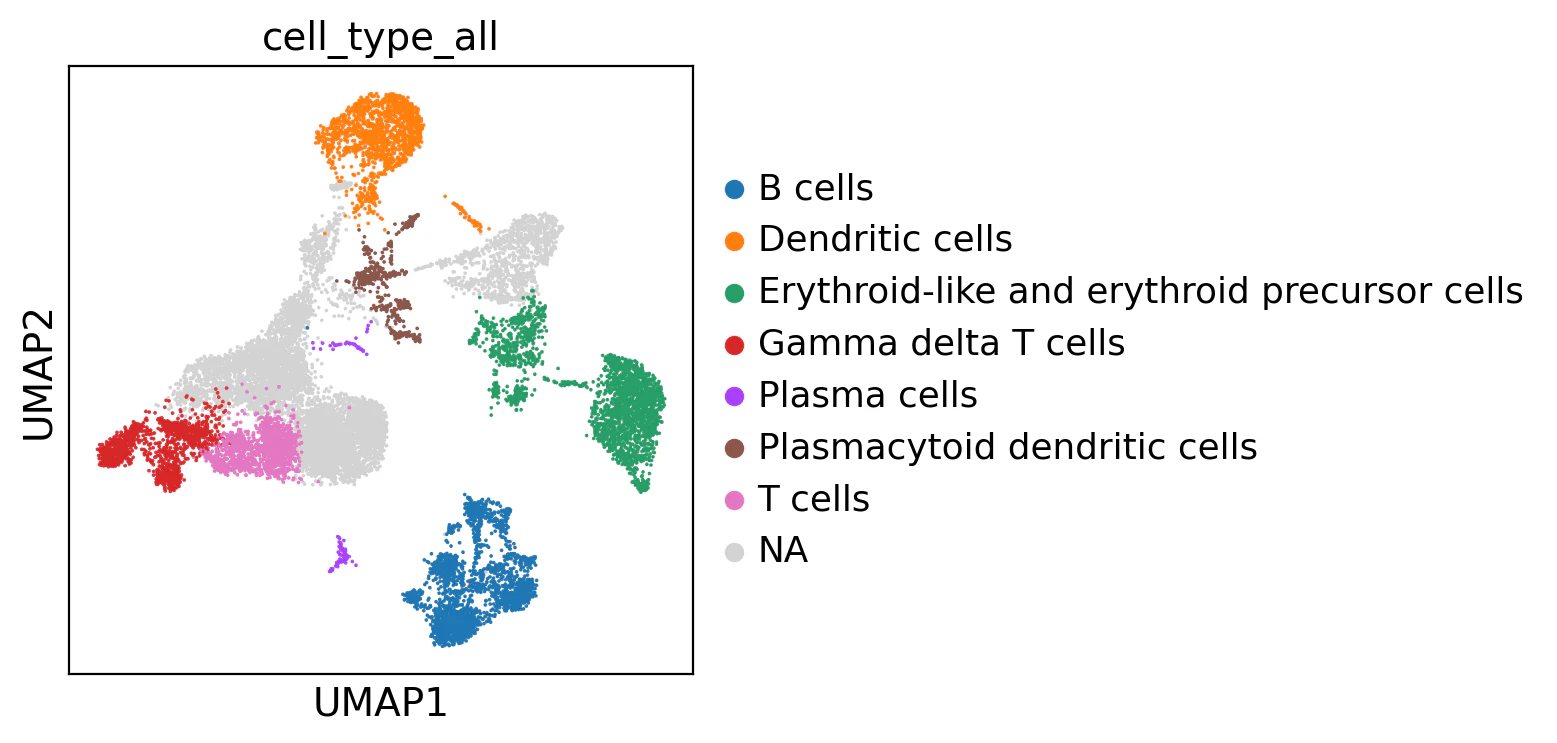
NAになってしまった箇所もあったので、PanglaoDBに含まれていない細胞マーカーがあることが分かりました。
また、差次的発現解析(DEGs)で得られた上位5つの遺伝子は、必ずしも細胞タイプの決定に直結するわけではないので発現している遺伝子を細かく見る必要があります。
よって、この割り当て方法は単純すぎて上手くいかないことが分かります、、
気を取り直して、別の手法も使ってみようと思います。
decouplerを使った自動アノテーション
decouplerの概要
decouplerは生物学データでいろいろなエンリッチメント解析を行うライブラリです。decouplerのチュートリアルに基づいて進めていきます。
decoupler Cell type annotation from marker genes
decouplerの適用方法
クラスタリングまで終わったデータを使います。
adata_clusterdにクラスタリング済みのデータが入っている前提で進めていきます。
マーカー遺伝子を取得
先ほども使ったPanglaoDBを使用します。
必要あれば再ロードしてください。
adata_decoupler = adata_clusterd.copy()
# PanglaoDB のデータセットを取得
markers = dc.get_resource(name="PanglaoDB", organism='human')
markers = markers[markers['human'].astype(bool) & markers['canonical_marker'].astype(bool) & (markers['human_sensitivity'].astype(float) > 0.5)]
# 不要な重複データを削除
markers = markers[~markers.duplicated(['cell_type', 'genesymbol'])]
ORA(過剰表現解析)を実行
公式ドキュメントによるORAの説明図です。

注釈をつけるとこのようになります。

つまり、有意に発現した遺伝子が、特定の遺伝子セットにどれだけ含まれるかを評価する手法であり、スコアが高いほど、その遺伝子セットが有意に過剰表現されていることを示しています。
# ORAを実行する
dc.run_ora(
mat=adata_decoupler,
net=markers,
source="cell_type",
target='genesymbol',
min_n=3,
verbose=True,
use_raw=False
)
adata_decoupler.obsm['ora_estimate']
実行すると、['ora_estimate']にそれぞれの細胞の-log10したP値が入っています。
# ORAのスコアを取得
acts = dc.get_acts(adata_decoupler, obsm_key='ora_estimate')
acts_v = acts.X.ravel() # 1次元配列に変換
max_e = np.nanmax(acts_v[np.isfinite(acts_v)]) #
acts.X[~np.isfinite(acts.X)] = max_e
acts
ここで登場したactsとは、AnnDataオブジェクトのような構造を持つdc.get_acts()の出力であり、acts.XにORAのスコアが行列形式で保存されています。
np.isfinite(acts_v)は有限値をフィルタリングするもので、np.nanmax()で最大値を取得しています。
次に、~np.isfinite(acts.X)により、NaNやinfがある位置だけTrueに変換しています。最後にそのTrueだけをmax_eに置き換えています。
NK細胞を抜き出してみます。
sc.pl.umap(acts, color=['NK cells', 'leiden_res_0.50'], cmap='RdBu_r')
sc.pl.violin(acts, keys=['NK cells'], groupby='leiden_res_0.50')


明確にクラスタ8におけるNK細胞のORAスコアが高いことが分かります。
では、そのORAスコアを使って、クラスターごとに細胞タイプのランキングを作ります。
# OEAスコアをもとにクラスターごとに細胞タイプのランキングを作る
df_decoupler = dc.rank_sources_groups(acts, groupby='leiden_res_0.50', reference='rest', method='t-test_overestim_var')
df_decoupler
ここのrefrenceはどのデータを比較の基準とするかを指定する引数であり、restは対象のクラスター以外のすべてのクラスターのスコアを比較することになります。
また、methodでは、分散を過大推定する t 検定(Welch’s t-test)を使用して、各クラスターと他のクラスターの間で、その細胞タイプが統計定期に有意に異なるかを評価します。
それぞれのクラスタの上位3位までの細胞タイプを取り出します。
n_ctypes = 3
ctypes_dict = df_decoupler.groupby('group').head(n_ctypes).groupby('group')['names'].apply(lambda x:list(x)).to_dict()
ctypes_dict
細胞タイプをリスト化した後に、クラスタをキー、細胞タイプをリストを値とする辞書ができます。

ヒートマップでクラスタとそれに対応する細胞リストのZスコアを可視化します。
sc.pl.matrixplot(acts,
ctypes_dict,
'leiden_res_0.50',
standard_scale='var',
colorbar_title='Z-scaled scores',
cmap='RdBu_r')
acts.Xはそのままだとスケールが違うので、standard_scale='var'で標準化します。

分布をプロットすることで個々の細胞タイプが分かります。
sc.pl.violin(acts, keys=['T cells', 'B cells naive','NK cells'], groupby='leiden_res_0.50')
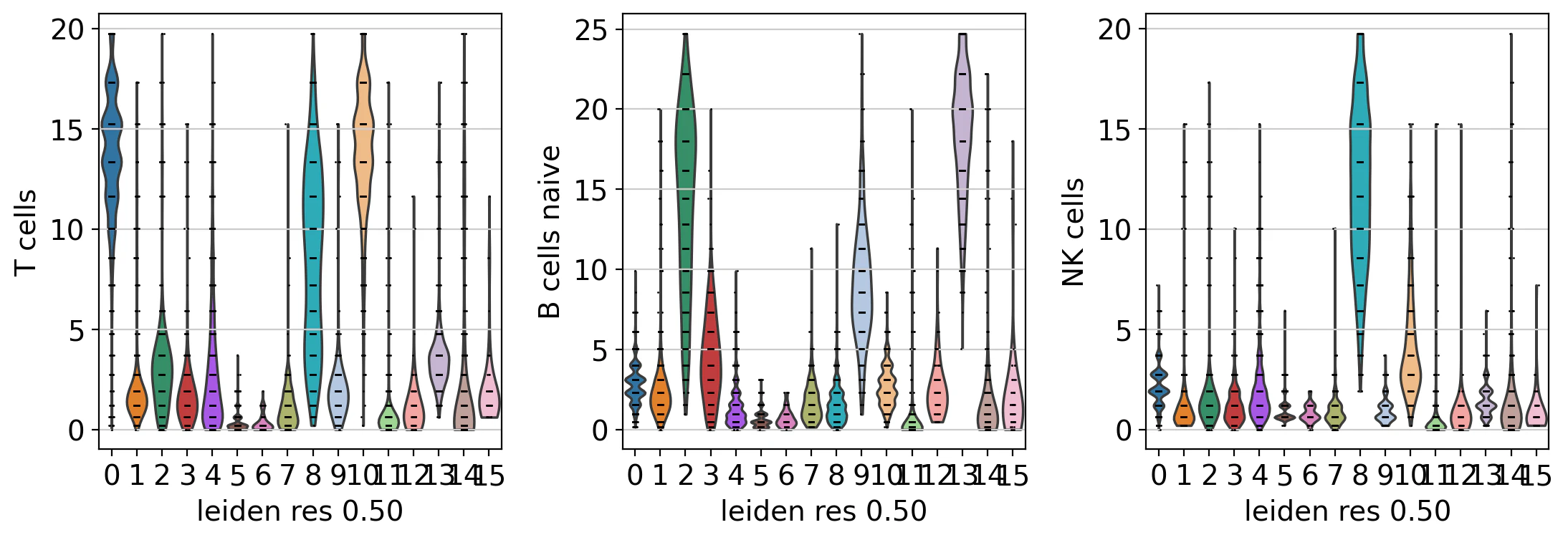
アノテーション
この後、エンリッチメントの結果に基づいて、手動で細胞タイプを決められますが、クラスターごとに予測された細胞タイプを割り当てることで自動予測が可能になります。間違いも発生しやすくなりますが、初手としては有用です。
# ランキング1位の細胞タイプを取り出す
annotation_dict = df_decoupler.groupby('group').head(1).set_index('group')['names'].to_dict()
annotation_dict

それでは、各クラスタ番号をキーにして、対応する細胞タイプを取得しそのラベルを格納します。
# アノテーション
adata_decoupler.obs['cell_type'] = [annotation_dict[clust] for clust in adata_decoupler.obs['leiden_res_0.50']]
sc.pl.umap(adata_decoupler, color='cell_type')
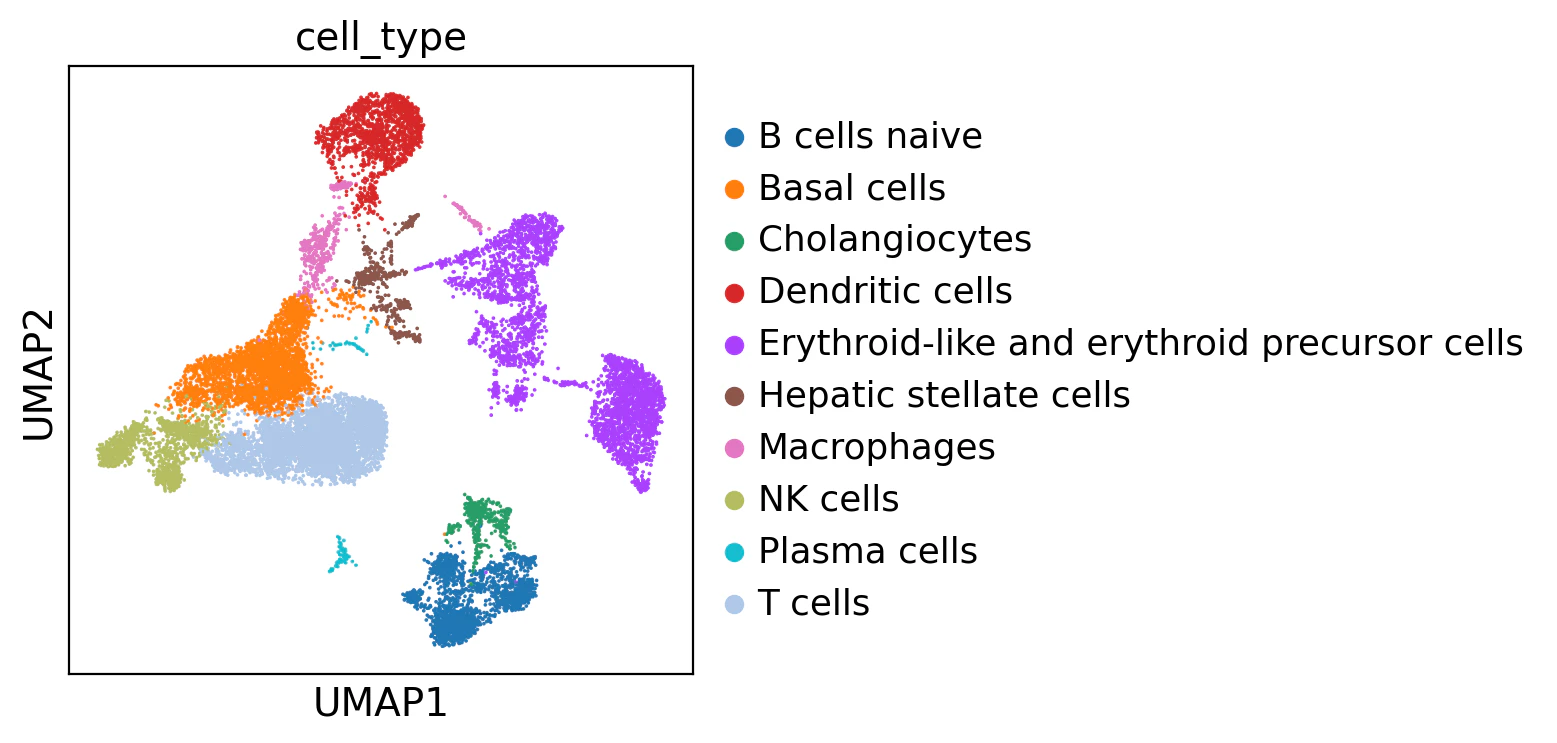
CellTypistを使った自動アノテーション
CellTypistの概要
シングルセルRNAシーケンス(scRNA-seq)データの細胞タイプを自動アノテーションするためのツールです。機械学習を活用し、事前に学習されたモデルを使って各細胞の分類を行います。
CellTypistの適用方法
Celltypistでは、スケーリング・次元削減前のデータを用いるため、事前にlayerへXの値を保存していました。
今回はそれを使っていきます。
# AnnDataのlayerに保存していたデータでadata.Celltypist.Xを上書きする
adata_CellTypist = adata.copy()
adata_CellTypist.X = adata.layers['before_DimRed'].copy()
取得可能なモデルの一覧を表示します。
# modelをアップデート
models.download_models(force_update = True)
models.models_description(on_the_fly = True)
今回は一番オーソドックスに使われる機械学習モデルを使用します。
# モデルを選択
model = models.Model.load(model = 'Immune_All_Low.pkl')
model

アノテーション
predictions = celltypist.annotate(adata_CellTypist, model = 'Immune_All_Low.pkl', majority_voting = True)
# 予測ラベルと信頼スコアを含むandataを取得
adata_CellTypist = predictions.to_adata()
adata.write('annotated_data.h5ad')
後の流れは同様です。
sc.tl.pca(adata_CellTypist, svd_solver='arpack')
sc.pp.neighbors(adata_CellTypist, n_pcs=20)
sc.tl.umap(adata_CellTypist)
sc.pp.neighbors(adata_CellTypist, n_pcs=10)
sc.tl.leiden(adata_CellTypist, resolution=0.5)
sc.pl.umap(adata_CellTypist, color='predicted_labels', title='UMAP with Annotations')

この結果は少々細かすぎるので、発現量が多く頻出する細胞タイプだけに絞って表示したほうが解釈できるようになります。
ちなみに、CellTypistを行うと、信頼スコアも手に入れられるため、このスコアが0.9以上の細胞ラベルのみをフィルタリングしてみます。
adata_high_conf = adata_CellTypist[adata_CellTypist.obs["conf_score"] >= 0.9]
# 高信頼スコアの UMAP を描画
sc.pl.umap(adata_high_conf, color="predicted_labels")
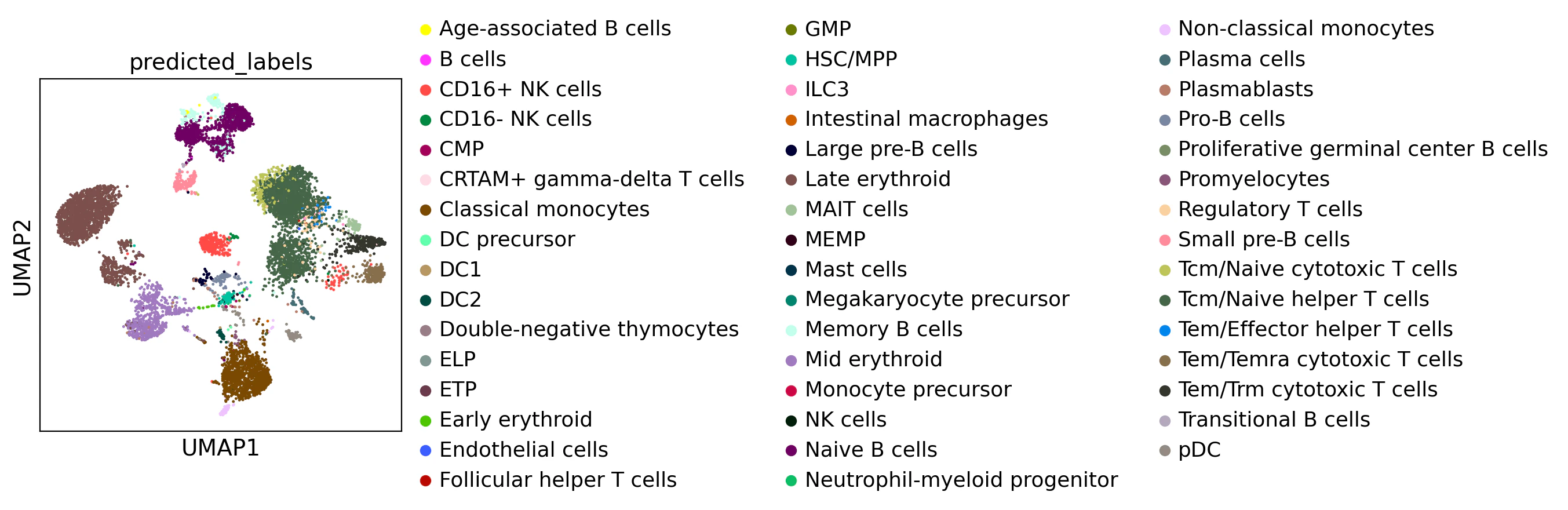
大きくは変わりません。現在使っていたモデルがかなり細かい分類まで行ってくれるものだったので、粗いモデルを使ってみます。
同じようにアノテーションした結果がこちらです。

信頼スコア0.9をフィルタリングしました。

NK細胞が見当たりませんね。
最後に、decouplerとCelltypistの結果を比べるために、クラスター8を抽出してみます。
クラスター8は、手動アノテーションでは、NK細胞かCD8+T細胞かがはっきりと判断できませんでしたが、decouplerではNK細胞に分類されていました。
adata_CellTypist.obs['celltypist_cell_type'] = adata_decoupler.obs['cell_type']
result = adata_CellTypist.obs[['leiden_res_0.50', 'celltypist_cell_type', 'predicted_labels']]
result

ここからクラスター8を抽出します。
result_8 = adata_CellTypist.obs.loc[adata_CellTypist.obs['leiden_res_0.50']=='8', ['celltypist_cell_type', 'predicted_labels']]
result_8
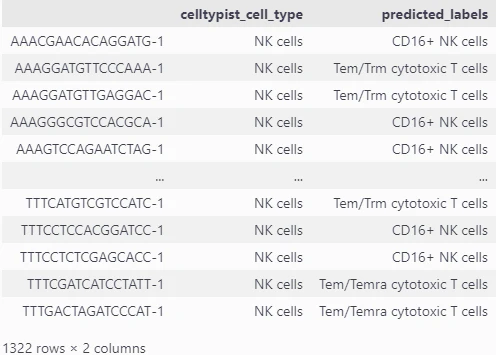
decouplerでは予測ラベルにNK細胞のほかにもT細胞が見受けられます。
この結果を可視化してみます。
adata_cluster_8 = adata_CellTypist[adata_CellTypist.obs["leiden_res_0.50"] == '8'].copy()
sc.pl.umap(
adata_cluster_8,
color='predicted_labels',
title = 'decoupler Annotation(Cluster8)'
)

クラスター8は大きくNK細胞群、T細胞群になっています。
decouplerのスコアリングでは、NK細胞群のほうが統計的に有意だったようです。
以上です。読んでいいただきありがとうございました。