はじめに
目的
タンパク質の配列-機能相関を調べる上で、
ある一つのタンパク質配列に対して任意のサイトに任意のアミノ酸変異を施した変異配列を
簡便に生成させたかったので作成しました。
pythonの練習も兼ねているので、他の方法やより良い方法など教えて頂けるとありがたいです。
GitHub
実行環境
jupyter notebook
python 3.7.4 (Anaconda)
pandas 0.25.3
biopython 1.74
内容
インポート
親となるタンパク質配列をfastaファイルで持っていると想定し、
ファイルの読み書きにbiopythonを使用。
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
import pandas as pd
関数定義
fastaファイルの配列名(id)と配列(seq)を辞書型で読み込み、
seq部分を1文字1列のDataFrameに変換した後、配列名の列をnameとして左端に挿入。
そのままではアミノ酸配列が0始まりとなってしまいサイト位置が直感とズレるので
1始まりに列indexを修正。
# fasta to dataframe
def fd(p):
d = {rec.id : list(str(rec.seq)) for rec in SeqIO.parse(p, "fasta")}
df = pd.DataFrame(d.values())
for i in d.keys():
name = i
df.insert(0, 'name', name)
df.iat[0, 0] = name
plus_one = {}
j = 1
for i in range(len(df.columns)+1):
plus_one[i] = j
j = j+1
df = df.rename(columns=plus_one)
return df
基本的な計算用の関数として、
特定の1サイトを特定のアミノ酸に変換するsdm()と
特定の1サイトを全てのアミノ酸に変換するssm()を作成。
# for calculation of site directed mutagenesis
def sdm(df, site, mut):
df_mut = df.copy()
df_mut.iat[0, 0] = df_mut.iat[0,0] + "_" + df_mut.iat[0,site] + str(site) + mut
df_mut.iat[0,site] = mut
return df_mut
# for calculation of site saturation mutagenesis
def ssm(df, site):
aa_list = ['R', 'H', 'K', 'D', 'E', 'S', 'T', 'N', 'Q', 'C', 'G', 'P', 'A', 'V', 'I', 'L', 'M', 'F', 'Y', 'W']
df_mut = df.copy()
for i in range(20):
df_mut = df_mut.append(df.iloc[0])
j = 1
for i in aa_list:
df_mut.iat[j, 0] = df_mut.iat[0,0] + "_" + df_mut.iat[0,site] + str(site) + i
df_mut.iat[j,site] = i
j = j + 1
df_mut.reset_index(drop=True,inplace=True)
return df_mut
複数の変異体をまとめて生成できるようにisdm()とissm()を作成。
これら2つの関数は任意の1箇所に変異が入った配列を個別の行に生成する。
# individual site directed mutagenesis
def isdm(df, site_list, mut_list):
mylist = []
j = 0
for i in site_list:
mylist.insert(j, sdm(df, i, mut_list[j]))
j = j+1
df = pd.concat(mylist)
df = df.drop_duplicates(subset='name')
df.reset_index(drop=True,inplace=True)
return df
# individual site site saturation mutagenesis
def issm(df, site_list):
mylist = []
j = 0
for i in site_list:
mylist.insert(j, ssm(df,i))
j = j+1
df = pd.concat(mylist)
df = df.drop_duplicates(subset='name')
df.reset_index(drop=True,inplace=True)
return df
複数箇所に同時に変異を入れた配列を生成するため、ssdm()を作成。
# simultaneous site directed mutagenesis
def ssdm(df, site_list, mut_list):
j = 0
for i in site_list:
df = sdm(df, i, mut_list[j])
j = j+1
return df
複数箇所の飽和変異の全組み合わせを生成するため、sssm()を作成。
# simultaneous site saturation mutagenesis
def sssm(df, site_list):
for x in range(len(site_list)):
df_mut = df.copy()
templist = []
j = 0
for i in range(len(df_mut)):
dftemp = ssm(df_mut[i:i+1], site_list[x])
templist.insert(j, dftemp)
j = j + 1
df = pd.concat(templist)
df = df.drop_duplicates(subset='name')
df.reset_index(drop=True,inplace=True)
return df
実行例
親配列サンプルとしてinput.txt(fasta形式、id:seq1 seq:MTIKE)を用意。読み込み。
# read fasta
file = "input.txt"
df_wt = fd(file)
df_wt

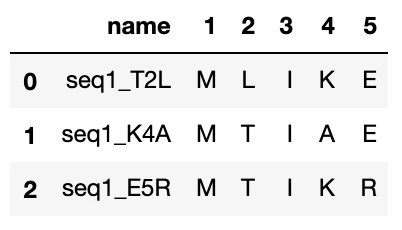
複数サイトの1変異体を生成したい場合↓
site_list = [2,4,5]
mut_list = ["L","A","R"]
df_isdm = isdm(df_wt, site_list, mut_list)
df_isdm
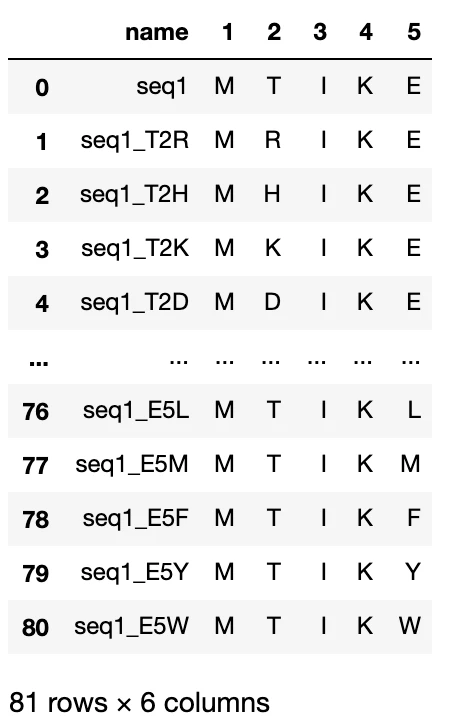
複数サイトの飽和変異体を生成したい場合↓
(例えば2番目から5番目までの飽和変異体)
site_list = range(2,6)
df_issm = issm(df_wt, site_list)
df_issm
複数サイトに変異を入れた一つの配列を生成したい場合↓
site_list = [2,4,5]
mut_list = ["L","A","R"]
df_ssdm = ssdm(df_wt, site_list, mut_list)
df_ssdm

複数サイトの飽和変異体の組み合わせを生成したい場合↓
site_list = [2,3]
df_sssm = sssm(df_wt, site_list)
df_sssm

配列の結合と重複削除
1文字1列のままだと研究上使いづらいので配列形式に結合し、同一配列を削除。
df = df_sssm.copy()
df.insert(1, 'sequence', "-")
df.loc[:,'sequence'] = df[1].str.cat(df[range(2,len(df.columns)-1)])
df_seq = df.iloc[:,0:2]
df_seq = df_seq.drop_duplicates(subset='sequence')
df_seq.reset_index(drop=True,inplace=True)
df_seq

エクスポート
csvとfastaにそれぞれ書き出し。
df_seq.to_csv("output.csv")
with open("output.txt", "w") as handle:
for i in range(len(df_seq.index)):
seq = Seq(df_seq.iloc[i,1])
rec = SeqRecord(seq, description="")
rec.id = df_seq.iloc[i,0]
SeqIO.write(rec, handle, "fasta")
おわりに
関数の作り方やfor文まわり等にとても無駄が多い気がするが、とりあえず得たい出力は得られるようになった。
今後の課題
- 単変異と飽和変異の混在("X"としたら飽和変異を実行)
- 複数の親配列の処理
- 巨大な配列の処理(実行時間短縮)
- コードの洗練