Qiime2による解析
本記事では、当研究室で細菌叢解析(16S rRNA V3-V4またはV4領域)を行う際に使用しているコマンドを記載しています。
使用するPCはMacですが、WindowsPCでもWSLでQiime2をインストール済みであれば、解析可能です。
Qiime2やRのインストール、WSLの使い方などは、すでに記事を書かれている方々がおられますのでそちらの記事を参考にして下さい。
1. Qiime2 (ver. 2025.4)の起動
conda activate qiime2-amplicon-2025.4
2. 作業ディレクトリの移動
cd Desktop/(~~.fastq.gzが入っている)フォルダ名
4のManifestFile作成で使用するコマンドは一番初めのアンダーバーで区切るようになっているため、fastq.gzを解凍する前にファイルの名前の変更が必要か確認しましょう。
(変更が必要な例)
サンプル名をA_1やA_2としてしまった場合、下記のようなファイルみとなります。
A_1_S1_R1.fastq.gz
A_1_S1_R2.fastq.gz
A_2_S1_R1.fastq.gz
A_2_S1_R2.fastq.gz
このままManifestFileを作成すると、sample-idの列は下記のようにAだけとなります。
sample-id,absolute-filepath
A,パス,forward
A,パス,reverse
A,パス,forward
A,パス,reverse
上記のようにならないために、解凍前にファイル名をA1_S1_R1.fastq.gzにしておけばA1とS1の間のアンダーバーで区切られるため問題なく次の作業に進むことができます。
3. .gzファイルの一括解凍
find ./ -type f -name "*.gz" -exec gunzip {} \;
4. ManifestFileの作成
ls *fastq | \
awk -F'_' -v P="$(pwd)" 'BEGIN { print "sample-id,absolute-filepath,direction" } \
{if(NR%2==0){print $1","P"/"$0",reverse"}else{print $1","P"/"$0",forward"}}' > ManifestFile.csv
5. demux.qzaの作成
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path ManifestFile.csv \
--output-path demux.qza \
--input-format PairedEndFastqManifestPhred33
6. demux.qzvの作成
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
7. demux.qzvの確認
qiime tools view demux.qzv
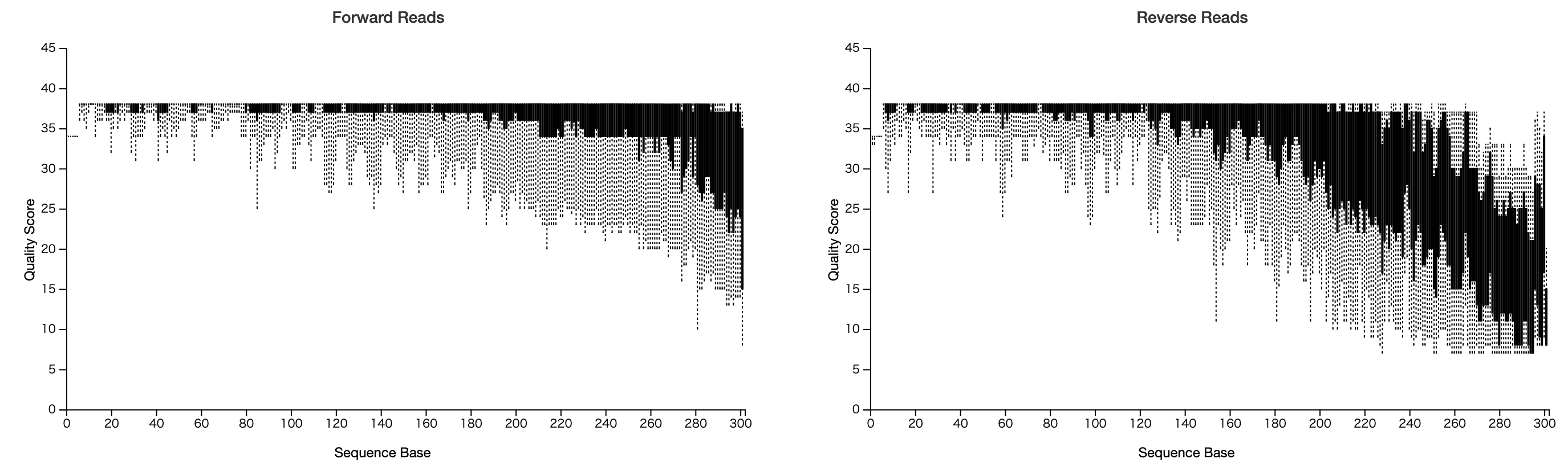
8. denoising作業(Bottom of BoxのQualityが20を超え、かつFとRの合計が500以上)
解析するデータ毎に図が異なりますが、例で示した図ではForwardが290あたり、Reverseが220あたりまでQualityが良いと判断できます。290+220=510で長さ的にも良いと判断しました。
qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux.qza \
--p-trunc-len-f 290 \
--p-trunc-len-r 220 \
--p-trim-left-f 0 \
--p-trim-left-r 0 \
--p-trunc-q 2 \
--p-n-reads-learn 1000000 \
--p-max-ee-f 2.0 \
--p-max-ee-r 2.0 \
--p-n-threads 10 \
--o-table table.qza \
--o-denoising-stats stats.qza \
--o-representative-sequences rep-seqs.qza \
--verbose
サンプル数が180件など多い場合はPCのスペックのせいでエラーが出る場合があります。その場合は、--p-n-threadsの値を10から1にしたり、--p-n-reads-learnを1000000から500000にしてみて下さい。
9. 塩基データrep-seqs.qzaから細菌分類データtaxonomy.qzaを作成
silva-138-99-515-806-nb-classifier.qzaはQiime2公式サイトからダウンロードできますが、MacPCのデスクトップ上のclassifierフォルダ内に入れてありますので、解析したいファイルがあるフォルダにコピペしてください。
V3-V4領域の解析では、silva-138-V3-V4-classifier-2025.4.qzaを使用し、
V4領域の解析では、silva-138-99-515-806-nb-classifier.qzaを使用します。
qiime feature-classifier classify-sklearn \
--i-classifier silva-138-V3-V4-classifier-2025.4.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
10. taxonomy.qzvの作成
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
11. 細菌分類データtaxonomy.qzaと存在量データtable.qzaを合わせて細菌の占有率グラフを作成
事前にSample IDとGroupやPeriodなどの情報を記載したタブ区切りのテキストファイルMetadata.txtを作成しておきましょう。Metadata.txtファイルは初めにエクセルで作成して保存するときにタブ区切りのテキスト形式で保存を選択すると良いです。
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file Metadata.txt \
--o-visualization taxa-bar-plots.qzv
####11-1. 植物由来のMitchondriaとChloroplastを除去
飼料や発酵食品などの細菌叢解析を進めているとMitochondriaやChloroplastがデータに入り込むことがあります。これはエラーである可能性が高いため、データから取り除く必要があります。既存のtable.qzaと区別するために、Qiime2公式サイトではtable-no-mitochondria-no-chloroplast.qzaと名付けているようです。
qiime taxa filter-table \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--p-exclude mitochondria,chloroplast \
--o-filtered-table table-no.qza
続く解析はtable.qzaをtable-no.qzaに変更するだけでOKです。
12. 系統樹の作成
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
13. 多様性指数(αとβ多様性の一部)の作成(chao1は作成されない)
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 10000 \
--m-metadata-file Metadata.txt \
--output-dir core-metrics-results
14. α希薄化曲線の作成(500readごとに10000readまで)
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 10000 \
--p-steps 21 \
--m-metadata-file Metadata.txt \
--o-visualization alpha-rarefaction.qzv
データのリード数によりますが、最大リード数が10000もありませんよというエラーが出たときは、そのエラーででてきた数字にするか、キリのいい数字にしたら良いと思います。
(例:9789だった場合は、--p-max-depth 9789とするか、--p-max-depth 9700にすれば良い)
15. chao1指数の計算
qiime diversity alpha \
--i-table table.qza \
--p-metric 'chao1' \
--output-dir core-metrics-results/chao1
--o-alpha-diversity core-metrics-results/chao1/chao1_vector.qza
なぜかchao1フォルダ内に指定した名称とは異なるファイル”alpha_diversity.qza”が生成されます。
16-1. α多様性指数の群間比較(shannon index, Kruskal-Wallis test)
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/shannon_vector.qza \
--m-metadata-file Metadata.txt \
--output-dir core-metrics-results/shannon
--o-visualization core-metrics-results/shannon/shannon_significance.qzv
16-2. α多様性指数の群間比較(chao1 index, Kruskal-Wallis test)
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/chao1/alpha_diversity.qza \
--m-metadata-file Metadata.txt \
--o-visualization core-metrics-results/chao1/chao1_significance.qzv
17-1. β多様性指数の群間比較(unweighted Unifrac, PERMANOVA)
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file Metadata.txt \
--m-metadata-column Group \
--o-visualization core-metrics-results/unweighted_unifrac_significance.qzv \
--p-pairwise
17-2. β多様性指数の群間比較(weighted Unifrac, PERMANOVA)
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/weighted_unifrac_distance_matrix.qza \
--m-metadata-file Metadata.txt \
--m-metadata-column Group \
--o-visualization core-metrics-results/weighted_unifrac_significance.qzv \
--p-pairwise
17-3. α多様性指数のraincloudプロットの作図(chao1 index)
--p-columnsの後にはMetadata.txt内で設定しているGroupやCategory、Periodなど任意の列の名前を入れましょう。
qiime stats alpha-group-significance \
--i-alpha-diversity core-metrics-results/chao1/alpha_diversity.qza \
--m-metadata-file Metadata.txt \
--p-columns Group Category \
--o-distribution core-metrics-results/chao1/dist.qza \
--o-stats core-metrics-results/chao1/stas.qza \
--o-raincloud core-metrics-results/chao1/chao1_raincloud_plot.qzv
17-4. α多様性指数のraincloudプロットの作図(shannon index)
qiime stats alpha-group-significance \
--i-alpha-diversity core-metrics-results/shannon_vector.qza \
--m-metadata-file Metadata.txt \
--p-columns Group Category \
--o-distribution core-metrics-results/shannon/dist.qza \
--o-stats core-metrics-results/shannon/stas.qza \
--o-raincloud core-metrics-results/shannon/shannon_raincloud_plot.qzv
α多様性については、このような図ができます。

Rによる作図
1. R Studioを起動
2. 作業ディレクトリを~~.qzaファイルが入ったフォルダに指定
3. ライブラリの起動
library(qiime2R)
library(phyloseq)
library(MicrobeR)
library(ggplot2)
4. データのインポート
otus <- read_qza("table.qza")
tree <- read_qza("rooted-tree.qza")
taxonomy <- read_qza("taxonomy.qza")
tax_table <- do.call(rbind, strsplit(as.character(taxonomy$data$Taxon), ";"))
colnames(tax_table) <- c("Kingdom","Phylum","Class","Order","Family","Genus","Species")
rownames(tax_table) <- taxonomy$data$Feature.ID
metadata <- read.table("Metadata.txt", sep='\t', header=T, row.names=1, comment="")
physeq <- phyloseq(otu_table(otus$data, taxa_are_rows = T), phy_tree(tree$data), tax_table(tax_table), sample_data(metadata))
5. 細菌占有率グラフの作成(Familyレベル)
Microbiome.Barplot(Summarize.Taxa(otus$data, as.data.frame(tax_table))$Family, metadata, CATEGORY="Group")
6. α多様性(Chao1 indexとShannon index)グラフの作成
colorやx、fillはMetadata.txtから最適なものを選択します。
p <- plot_richness(physeq, color = "Period", x = "Group", measures=c("Chao1", "Shannon"))
p <- p + geom_boxplot(aes(fill = Period), alpha=0.3) + theme_bw() + theme(text=element_text(size=20), axis.text.x=element_text(angle=45, size=15, hjust=1), axis.title.x = element_blank(), axis.title.y = element_blank()) + scale_x_discrete(limits=c("X軸1番目", "X軸2番目", "X軸3番目"))
plot(p)
7-1. β多様性(unweighted UniFrac, PC1vs PC2)グラフの作成
α多様性のときと同様、colorやshapeなどはMetadata.txtから最適なものを選択します。
凡例はアルファベット順や先頭の数字の少ない順に自動的に並びますので、〜_〜_discreteで並び替えます。例えば、Control → Protein → Fiberと並べたいのにデフォルトでは、C → Fiber → Proteinとアルファベット順に並んでしまいます。また、B5 → B10 → B20と濃度の低い方から並べたいのにデフォルトでは、B10 → B20 → B5というように先頭の数字でソートされてしまいます。このような時にdiscreteは役に立ちます。プロットの色も変えたい場合はscale_color_discreteをscale_color_manualに変えて、value=c("変えたい色1","色2","色3")をlimits=c()の横に加えてください。
ord1 <- ordinate(physeq, method = "PCoA", distance = "unifrac")
p1 <- plot_ordination(physeq, ord1, color = "Period", shape = "Group")
p1 <- p1 + geom_point(size=4) + theme_bw() + ggtitle("PCoA + UnWU")+scale_shape_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目"))+scale_color_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目")) + theme(text=element_text(size=15))
plot(p1)
7-2. unweighted UniFracのPC2 vs PC3グラフの作成
p1 <- plot_ordination(physeq, ord1, color = "Period", shape = "Group", axes = 3:2)
p1 <- p1 + geom_point(size=4) + theme_bw() + ggtitle("PCoA + UnWU")+scale_shape_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目"))+scale_color_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目")) + theme(text=element_text(size=15))
plot(p1)
7-3. β多様性(weighted UniFrac, PC1vs PC2)グラフの作成
ord2 <- ordinate(physeq, method = "PCoA", distance = "wunifrac")
p2 <- plot_ordination(physeq, ord2, color = "Period", shape = "Group")
p2 <- p2 + geom_point(size=4) + theme_bw() + ggtitle("PCoA + WU")+scale_shape_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目"))+scale_color_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目")) + theme(text=element_text(size=15))
plot(p2)
7-4. weighted UniFracのPC2 vs PC3グラフの作成
p2 <- plot_ordination(physeq, ord2, color = "Period", shape = "Group", axes = 3:2)
p2 <- p2 + geom_point(size=4) + theme_bw() + ggtitle("PCoA + WU")+scale_shape_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目"))+scale_color_discrete(limits=c("凡例1番目", "凡例2番目", "凡例3番目")) + theme(text=element_text(size=15))
plot(p2)
α多様性やβ多様性の作図ではプロットや文字の大きさ・角度・色などは個人的な好みもあろうかと思います。
適宜、調整してください。
##STAMP(statistical analysis of taxonomic and functional profiles)による解析
細菌の占有率の棒グラフはGenusレベルになるとパッと見で試験区間の違いを正確に理解することはできません。
試験区間でどのような菌の占有率が違うかをより分かりやすく表示する際に当研究室ではSTAMPを使用しています。
STMAPには2つのファイル(Profile fileとGroup metadata file)が必要になります。
STAMPは長らく更新されていませんので、Qiime2で得られるqzaファイルから直接STAMP用ファイルを作成することは難しいです。
ここまでコードを入力すればファイルが作成されていましたが、STAMP用ファイルの作成はかなり原始的ですのでスクショと共に方法を記載します。
#####Profile fileの作成方法
-
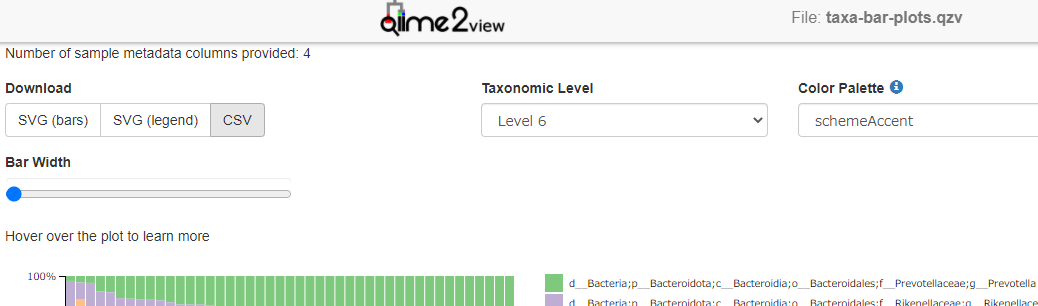
Qiime2で作成したtaxa-bar-plots.qzvファイルをQiime2 viewで閲覧し、Level 6のCSVファイルをダウンロードします。なお、Level7までCSVファイルを得られますが、シーケンスのリード数やデータベースのバグ?によりSTAMP上でエラーが度々生じますので、Level6までで留めておいたほうが良いです。
-
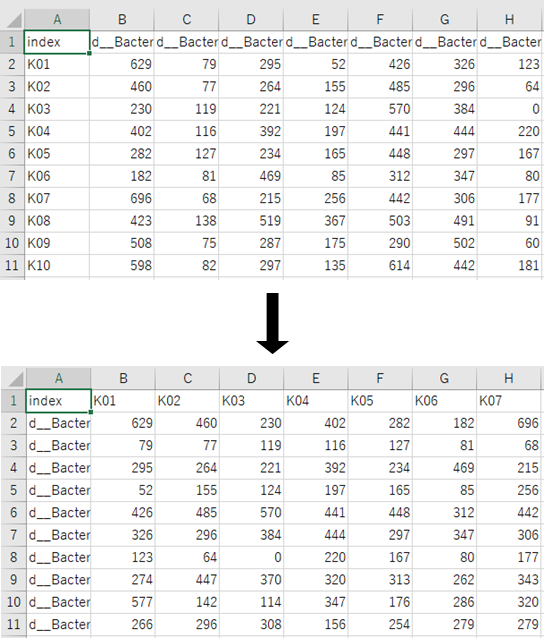
ダウンロードしたCSVファイルをエクセルで開き、行と列を入れ替えます。
-
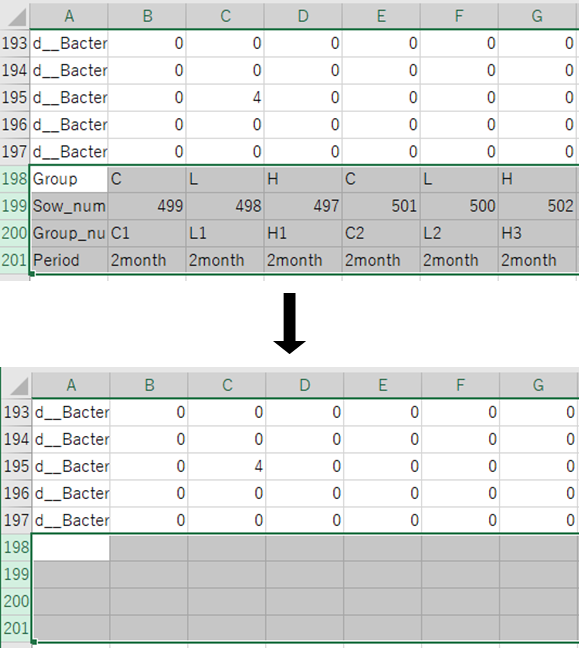
不要な行(Metadata.txt由来のデータ)を削除します。
-
1列目(細菌の名称)をKingdomからGenusまで区切ります。
菌の名称の前にあるアルファベット1文字(dはDomain、pはPhylum、gはGenus)の直後にアンダーバー2個があります。
またDomainとPhylumの間といったLevelの間にはセミコロンがあります。
これらをエクセルのデータタブの区切り位置に指定して区切ります。
なお、そのままアンダーバーを区切り位置に指定すると、~~~_groupといった名称の菌が名称の途中で区切られるため、アンダーバー2個はハテナマーク(?)にすべて置換し、?を区切り位置に指定すると良いでしょう。
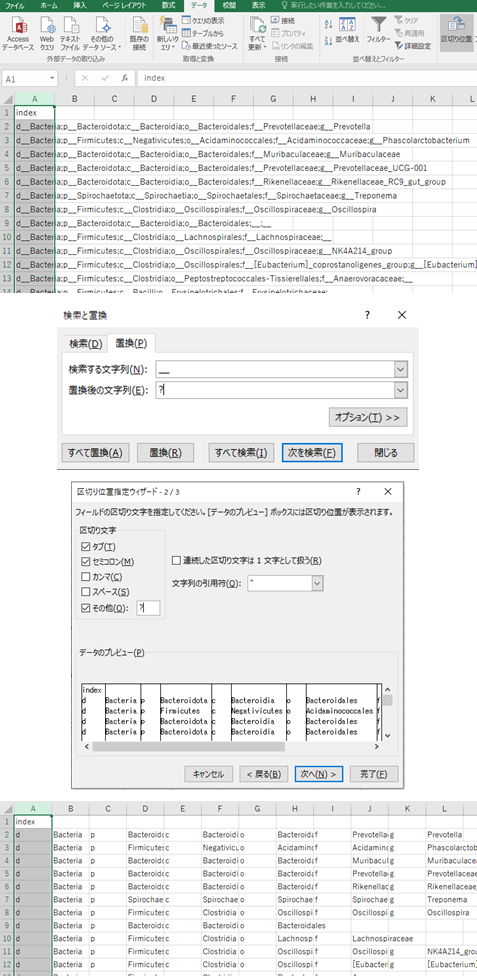
-
A、C、E、G、I、K列を削除し、1行目にKingdom、Phylum、Class、Order、Family、Genusと加筆します。
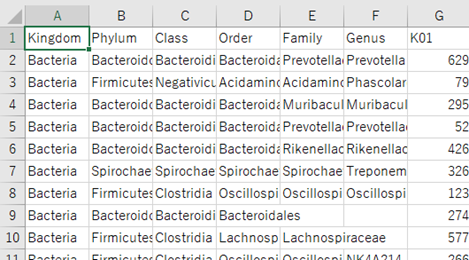
-
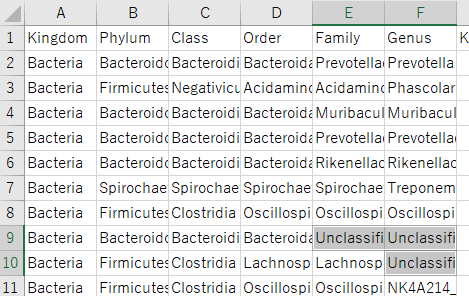
AからF列の空欄をUnclassifiedに置換し、タブ区切りのテキストファイルで保存します。STAMP_profile.txtといった名前にするとわかりやすいです。
#####Group metadata fileの作成方法
- Qiime2で使用したMetadata.txtをエクセルで開き、1行目、1列目のsampleidをSample IDに変換し、タブ区切りのテキストファイルで保存します。STAMP_metadata.txtといった名前にするとわかりやすいです。
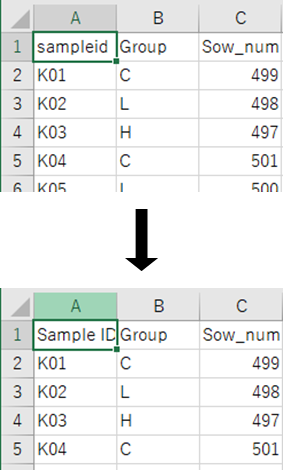
#####STAMPへのデータのインポート方法
STAMPを起動し、上記で作成した二つのファイルをLoadします。
なお、STAMPは日本語を認識できないため、フォルダ名に日本語があると、そこに入れられたファイルは認識できません。また、外付けHDDといったデスクトップと離れたディレクトリにファイルがあるのもダメなようです。デスクトップ上に英語のみのフォルダ(例えば、STAMP)を作成し、そこにファイルを入れておきましょう。
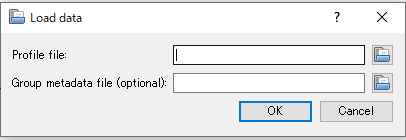
あとは試験区間の比較やグラフの微調整をソフトで行えばOKです。
間違いや改善点等がありましたらご連絡頂けますと幸いです。