はじめに
Bioinformaticianになるつもりはないが、仕事の関係上雰囲気だけでも理解しておきたい、ということで次世代シークエンサー DRY解析教本 を始めた。
Level2-2 発現解析まで辿り着いたものの、2019年現在 deprecatedなコマンドが存在しており、回避策が必要である。また、予備知識を残しておきたい気持ちもあり、メモを残すことにした。
解析準備
homebrewでインストールできるものはhomebrewでインストールする。
brew tap brewsci/science
brew tap brewsci/bio
brew install fastqc
brew install sratoolkit
brew install samtools
tophatは depreacated なので後継の hisat2 を使う。
brewからインストールを試みるが、Hisat2がerrorと同様の問題にぶち当たったので、大人しくバイナリをインストールする方針に切り替える。
wget http://ccb.jhu.edu/software/hisat2/dl/hisat2-2.1.0-OSX_x86_64.zip
unzip hisat2-2.1.0-OSX_x86_64.zip
cufflinksもsegmentation faultになるので、OS Xのバイナリをインストールする。
wget http://cole-trapnell-lab.github.io/cufflinks/assets/downloads/cufflinks-2.1.1.OSX_x86_64.tar.gz
gunzip -c cufflinks-2.1.1.OSX_x86_64.tar.gz | tar xopf -
あとは適当なディレクトリに置いて、PATH を通しておけばよろしい。
データの入手
レファレンス配列データの入手
wget http://shujunsha.com/NGS_DAT/Lv2_2/iGenome/Homo_sapiens/NCBI/build37.2/Sequence/Bowtie2Index/genome.fa
wget http://shujunsha.com/NGS_DAT/Lv2_2/iGenome/Homo_sapiens/NCBI/build37.2/Annotation/Archives/archive-2014-06-02-13-47-29/Genes/genes.gtf
1つ目はGene Feature Format(GTF)で、遺伝子アノテーションを記述しているファイルである。
featureはexon, intron, miRNAなどを指しており、染色体やstart/end positionなどが記述されている。
2つ目はヒューマンゲノムのリファレンス配列。
解析データの入手
今回は論文等で公開されているデータからピックアップ。もちろん、自分たちが実験で次世代シークエンサから得たデータを使うこともある(はず)。
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/sralite/ByExp/litesra/ERX/ERX182/ERX182652/ERR266335/ERR266335.sra
fastq-dump ERR266335.sra
これでSRAファイルからFASTQファイルが得られる。
で、中身はと言うと...
$ head -n4 ERR266335.fastq
@ERR266335.1 HWI-ST628_0112:1:1101:1233:2169#0 length=100
CTTCAATAGGATGGCTGTTATCTAAGGCGTCAAGCTCCTGGGCACGGGTGTAATCAGCAGAAACAAACTGAGTCCAAATATCATACTCTGGGAAGCAGTG
+ERR266335.1 HWI-ST628_0112:1:1101:1233:2169#0 length=100
CCCFFFFFHHHHHJJJJJJIJIIJJJJJJHIJJJJJJJJJJJJJJJJJ@GBHIJJJHHHHHFFFFFEDEEEDCDDDDCCDFEEEDDDDDDDD?C?B?CAC
NGS解析基礎[1]等によれば、
1行目: ヘッダ
2行目: リードの塩基配列
3行目: ヘッダ
4行目: リードのクオリティ
とのこと。
解析
クオリティチェック
fastqc --nogroup -o ./output/dir/ ERR266335.fastq
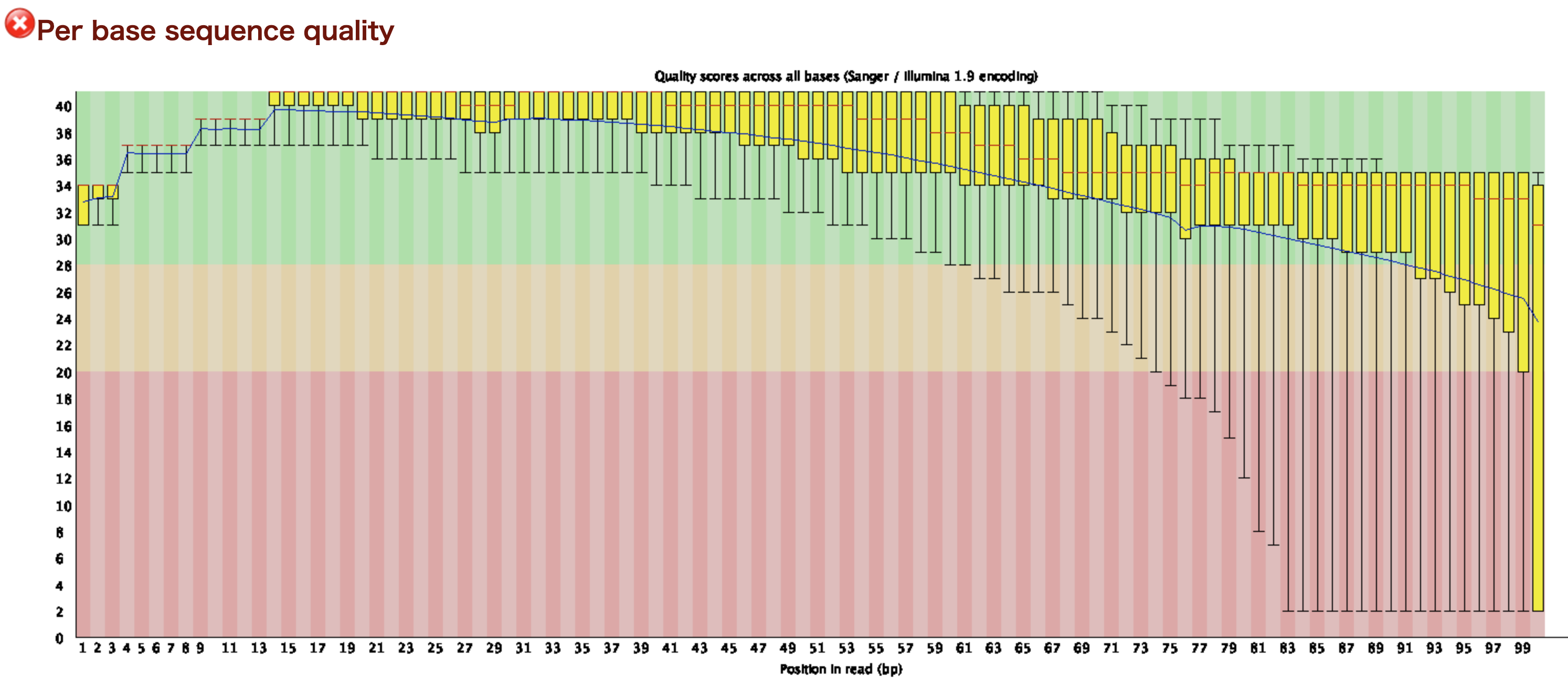
横軸はリード⻑、縦軸はquality valueを表す。リードの位置における全体のクオリティの中央値や平均を確認できる。⾚線は中央値、⻘線は平均値、⻩⾊のボックスは25%〜75%の領域を表す。上下に伸びた⿊いバーが10%〜90%の領域を意味する。 [2]
一般に3'末端に行くほどクオリティスコアが悪くなるらしい[3] 。5'→3'方向にシークエンシングしていくのでエラーが累積していくということらしいが...シークエンスの原理が分かれば納得できるようになるのだと思う。
トリミング
fastq_quality_trimmer -Q 33 -t 20 -l 30 -i ERR266335.fastq -o ERR266335_trim.fastq
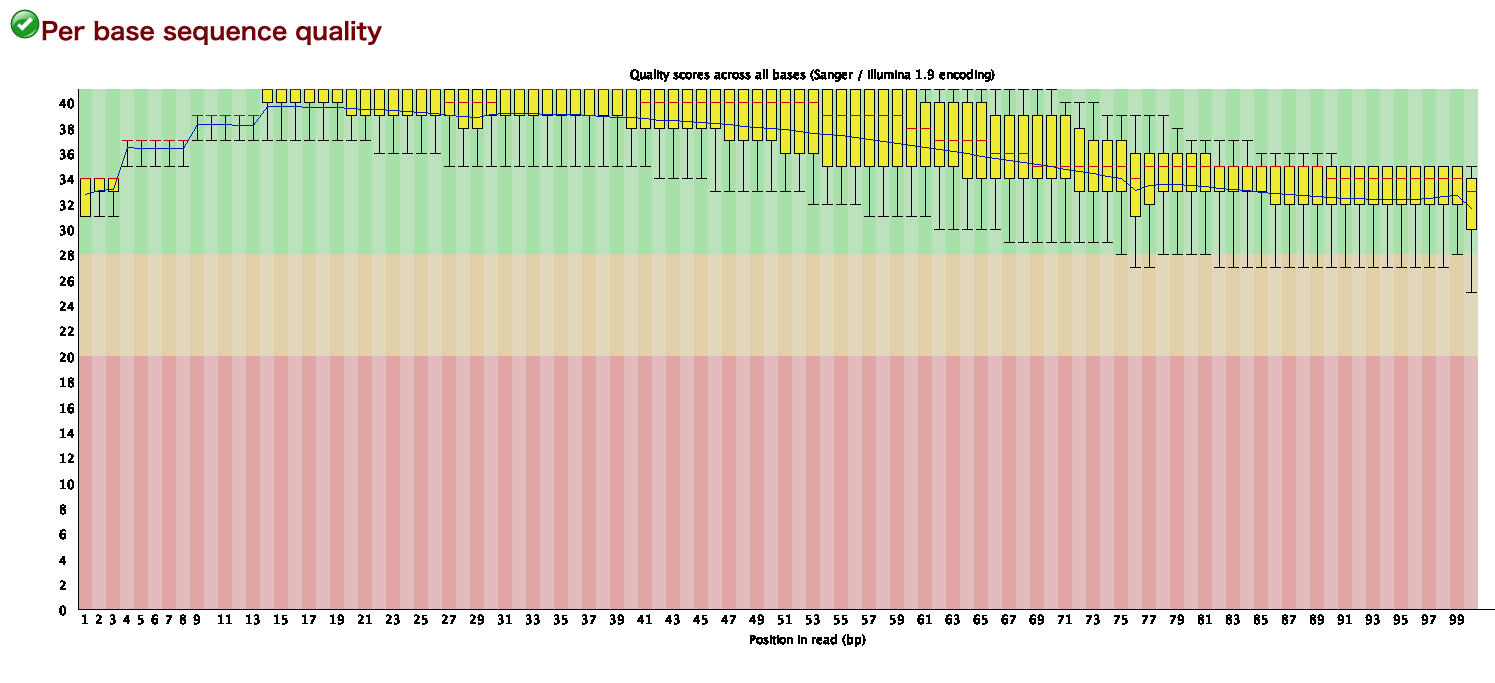
3'末端のクオリティのバラつきは減っていて、確かにクオリティスコアが20以上になってる。
マッピング
HISAT2を利用するので、DRY解析教本には書かれていない部分。
サルマップ2018 (1) 爆速Aspera Connect & HISAT2などを参考にさせてもらいながら、試行錯誤しながらやる。
まず、インデックスを作成する。
hisat2-build genome.fa genome_index
次いで、HISAT2でマッピングをする。
後続の処理でcufflinksを使うので、dta-cufflinksオプションを指定するのがミソ。
初回はこのオプションを指定しなかったので、BAM record error: found spliced alignment without XS attribute が出て困った。Pythonバイオ/ツール/RNAseq④発現解析(続き) に解決策が書いてあり、大変助かった。
hisat2 -p 4 --dta-cufflinks -x genome_index -U ERR266335_trim.fastq -S ERR266335.sam
BAMファイルを作成する。
samtools sort -@ 4 -O bam -o ERR266335.sort.bam ERR266335.sam
ようやく遺伝子発現量の解析ができる。
cufflinks -p 4 -g genes.gtf ERR266335.sort.bam -o output/
マージ
ERR266335以外の他5件についても同様の処理を行って、いい感じにマージする。
ls output/ERR266*/transcripts.gtf > transcripts.gtf.txt
output/ERR266335/transcripts.gtf
output/ERR266337/transcripts.gtf
output/ERR266338/transcripts.gtf
output/ERR266347/transcripts.gtf
output/ERR266349/transcripts.gtf
output/ERR266351/transcripts.gtf
cuffmerge -p 2 -o output/fpkm_compare -g references/genes.gtf -s references/genome.fa transcripts.gtf.txt
サンプル間のFPKMを比較
FPKMで正規化された遺伝子発現量を得る。
cuffdiff -p 2 --upper-quartile-norm -o output/cuffdiff_result/ -L ERR266335_P0,ERR266349_P2,ERR266337_P5,ERR266351_P7,ERR266338_P10,ERR266347_Adult output/fpkm_compare/merged.gtf output/ERR266335/ERR266335.sort.bam output/ERR266349/ERR266349.sort.bam output/ERR266337/ERR266337.sort.bam output/ERR266351/ERR266351.sort.bam output/ERR266338/ERR266338.sort.bam output/ERR266347/ERR266347.sort.bam
RでDendrogramの描写
R Studio(version1.2.1335)でコマンドを実行する。
BiocManager::install("cummeRbund")
library('cummeRbund')
setwd("~/sandbox/genomics/NGS/level2_2/output/cuffdiff_result/")
extdataPath <- paste("~/sandbox/genomics/NGS/level2_2/output/cuffdiff_result/", sep = " ")
cuff <- readCufflinks(extdataPath)
my.genes <- genes(cuff)
par(mar = c(8, 5, 5, 1))
dend <- csDendro(my.genes)
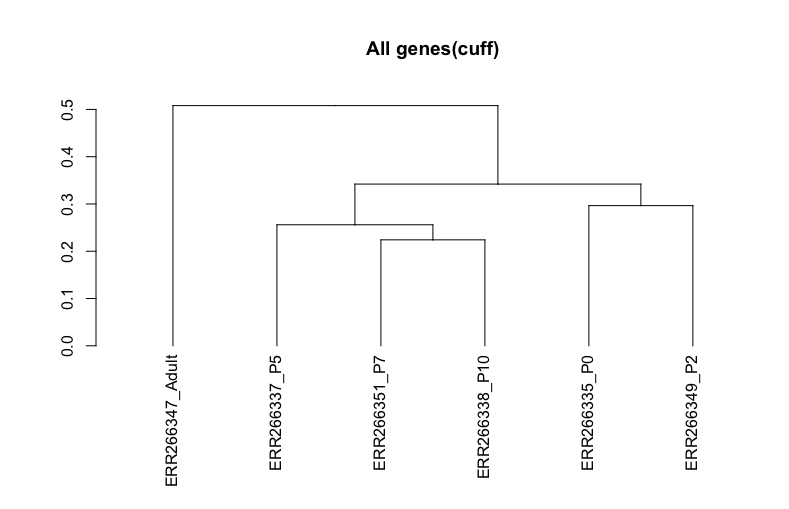
次世代シークエンサー DRY解析教本と同様のクラスタが図示されたので、これで良さそう。
まとめ
brewでインストールできるツールはbrewでインストールし、どうにもならないものはOS Xのバイナリを利用して、2019年現在も動く環境を用意できた。
実験の再現性等を考えるとDockerを選択するのが良い、という印象。
書籍のコンセプトの都合上、そうできなかったのは理解している。
参考資料
本文中でリンクしてない参考資料は以下に掲載。
- [1] amelieff. “NGS解析基礎”. https://biosciencedbc.jp/gadget/human/20150803and26_yamaguchi.pdf
- [2] amelieff. “平成28年度NGSハンズオン講習会 RNA-seq解析”. https://biosciencedbc.jp/gadget/human/20160727_amelieff_20160803.pdf
- [3] biopapyrus. “クオリティコントロール”. https://bi.biopapyrus.jp/rnaseq/qc/