ヒトの体は37兆個の細胞からできています。細胞は23000個の遺伝子をもっており、この遺伝子に突然変異が積み重なって、無制限に増殖するようになったものががん細胞と考えられています。
このページでは、Rubyを使ってcBioPortalから遺伝子情報を取得します。次に、得られたデータをRumaleというライブラリを使用して簡単な機械学習(Random Forest)にかけてみます。
cBioPortalとは
アメリカのTCGA(The Cancer Genome Atlas)というグループが、多くのがん細胞について,ゲノム、メチル化異常、遺伝子、タンパク質発現異常についての解析結果を、cBioPortalというサイトで結果を公開しています。特に制限はなくログイン不要で誰でもアクセスできます。
ブラウザでデータを眺めてもよいのですが、今回はJupyter + Rubyを用いてデータを取得することにします。
Rubyとは

やわらかい書き味に定評があるプログラミング言語です。
WebAPIとは
WebAPIは、Webサービスをプログラミングから操作するための方法です。インターネットからプログラムをつかって情報を取得できる仕組み、というぐらいの理解です…。
https://www.cbioportal.org/api/swagger-ui.html
こちらがcBioPortalのAPIのリファレンスです。これを常時参照します。
Rumaleとは
Rubyで定番の機械学習ライブラリです。
https://github.com/yoshoku/rumale
KEGGとは

KEGGは京都大学が作成した世界的に有名なバイオインフォマティクス用のデータベースです。多くの疾患に関連するパスウェイ(遺伝子やタンパク質の相互作用を経路図)などが登録されています。
目標
今回は簡単に実行できそうで、かつ少しは意味が有る結果が得られそうな
- KEGGに登録された胃癌に関連する遺伝子のmRNA発現量から、リンパ節転移の有無を予想することはできるか?
としてみました
前準備
Jupyter + Ruby 環境を導入します。導入方法は少々面倒なところがあります。インターネット上の各種資料を参照してください。やり方によっては、Google Colab でRubyを動かすことも可能です。サイドパネル等でリファレンスを表示しておくと便利だと思います。
必要なライブラリをインストールします。
# GitHubのリポジトリからgemをインストールするためのツール
gem install specific_install
# データフレーム
gem specific_install https://github.com/SciRuby/daru
# 機械学習
gem install rumale
# Httparty → Daru 変換用
gem install daru-apiclient
ライブラリーを読み込みます
require 'daru/apiclient'
require 'awesome_print'
require 'rumale'
がんのゲノムデータを集めたcBioPortalのAPIに接続する準備をする
クライアントを作成します。Daru::APIClientは、HTTPartyで取得したjsonをDaru::DataFrameに変換するだけの自作の超薄いツールです。cBioPortalを利用するために作りました。好みのクライアントがある方は使わなくても大丈夫です。
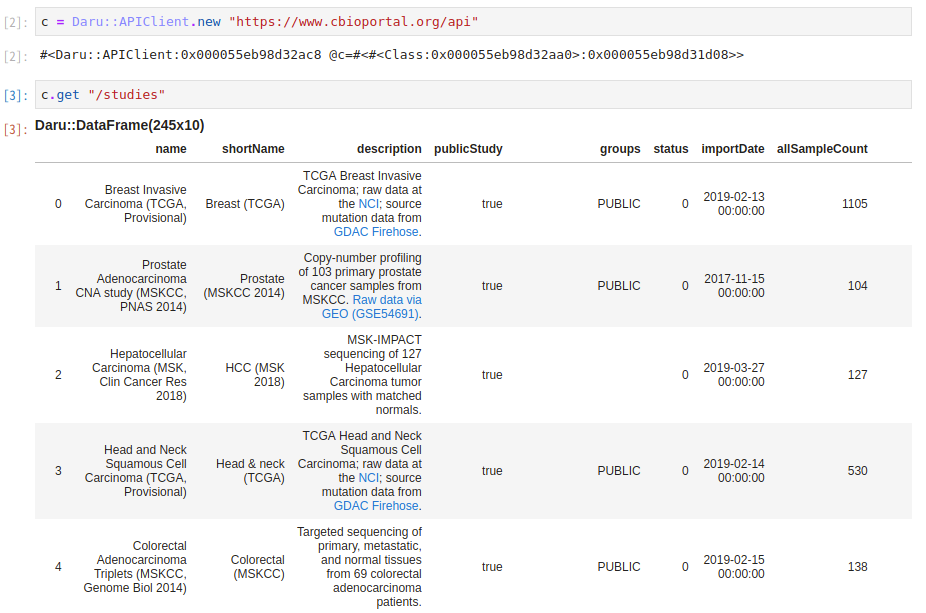
c = Daru::APIClient.new "https://www.cbioportal.org/api"
ためしに
c.get "/studies"
とタイプして、cBioPortalのスタディの一覧が表示されればうまくいっているはずです。
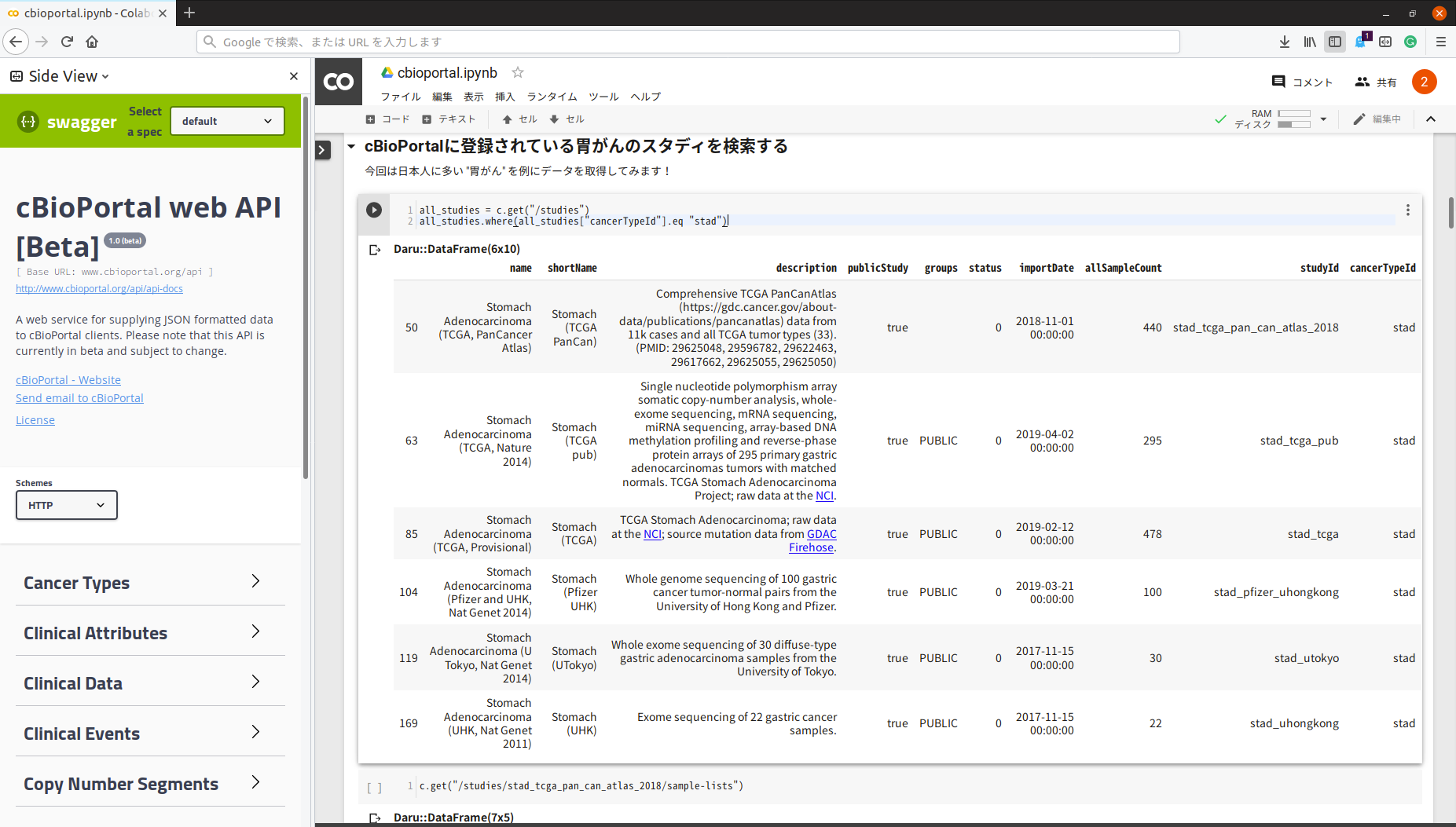
cBioPortalに登録されている胃がんのスタディを検索する
胃がんのスタディのリストを表示します。stad は 胃癌(Stomach adenocarcinoma)を示すIDです。スタディとは臨床研究の単位を表す単語です。
studies = c.get("/studies")
studies.where(all_studies["cancerTypeId"].eq "stad")
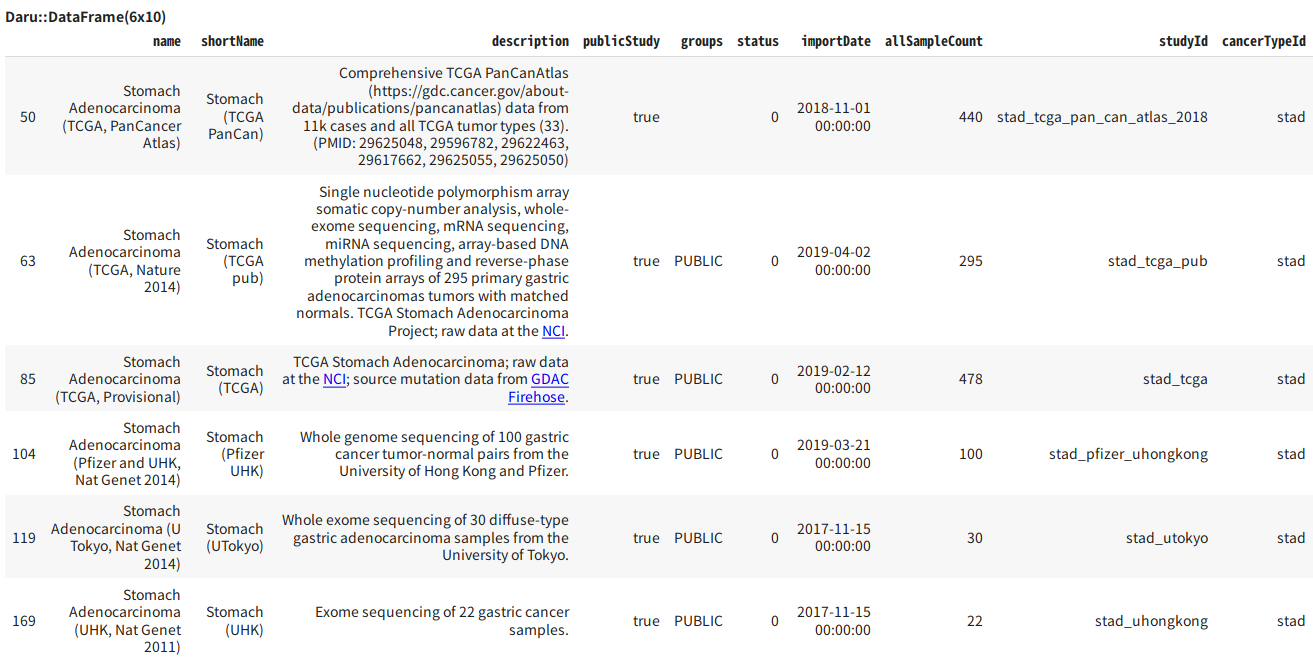
上から5番目のデータは東京大学のデータですね。日本のデータも登録されていることがわかって少しうれしくなりますね。しかし、ここではサンプル数が多く、更新日も新しいTCGAのデータ(stad_tcga)を使用することにします。
スタディの詳細をもっと見たい場合は
ap c.hget("/studies/stad_tcga")
とするともう少し詳しいサンプル数などが表示されます。

スタディに登録されているサンプルリストを確認する
あとでIDが必要になるので、先にスタディに登録されているサンプルリストを取得しておきます。
c.get("/studies/stad_tcga/sample-lists")
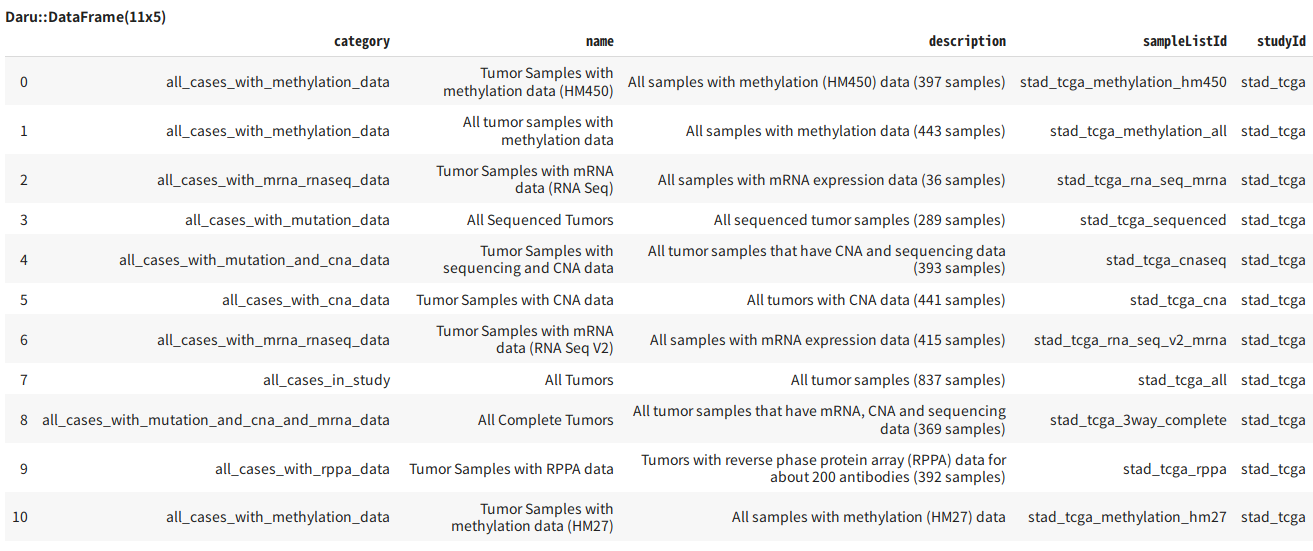
今回はmRNAの発現量を扱うので、必要なsampleListIdは、stad_tcga_rna_seq_v2_mrna ですね。
スタディーの中でアクセスできるMolecular Profilesのリストを取得する
あとで必要になるので、スタディに登録されている分子プロファイルのリストを取得しておきます。
c.get("/studies/stad_tcga/molecular-profiles")
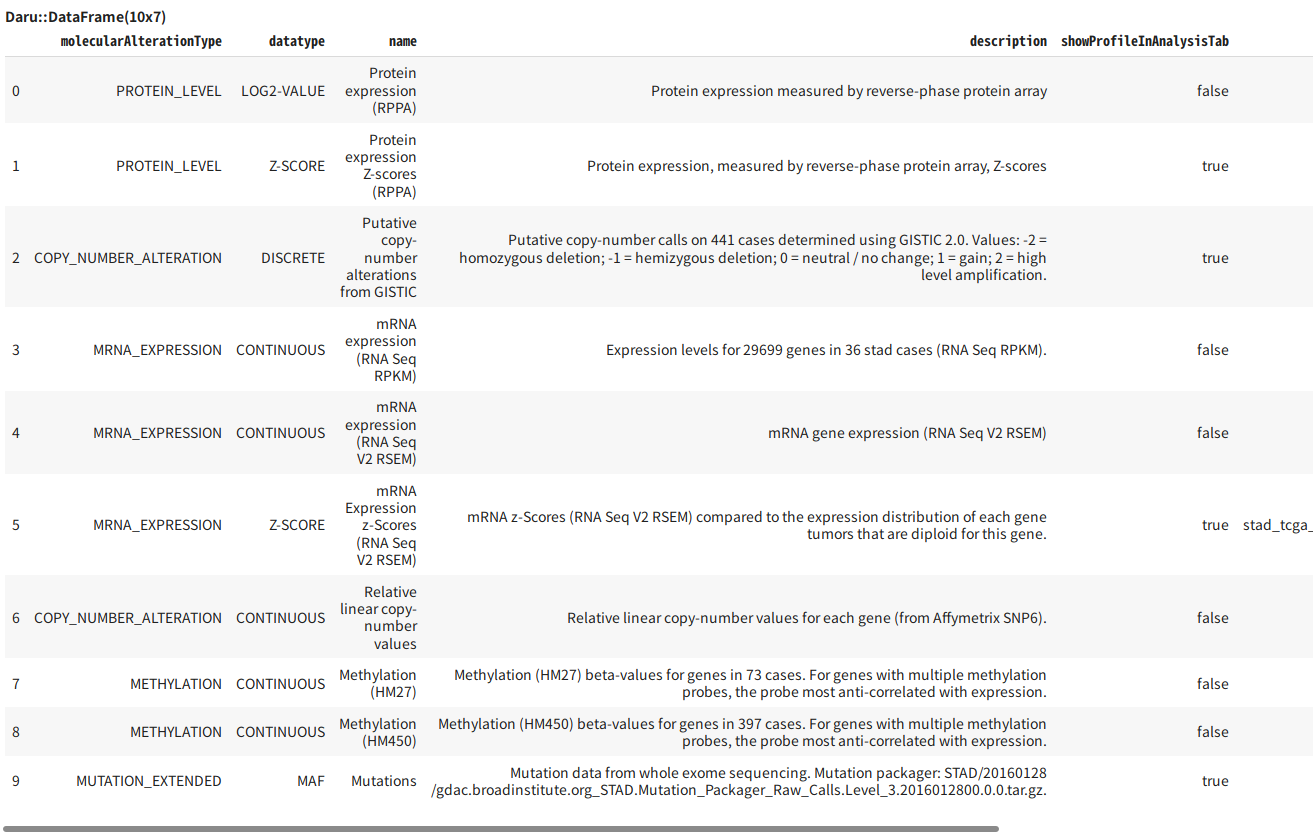
今回はmRNAの発現量を扱うので、stad_tcga_rna_seq_v2_mrna を使ってみます。例えばZスコアを使う場合は stad_tcga_rna_seq_v2_mrna_median_Zscores となります。
TogoWS 経由で 胃がん のパスウェイに関わる遺伝子のEntrez Gene ID のリストを取得する
全ての遺伝子のデータを使ってリンパ節転移の有無を予想するモデルを作成すると、サンプル数(400程度)よりも遺伝子(20000程度)の方がずっと多いということになり、得られた結果の意味がなくなってしまうかもしれません。
また、今回はすでにバイオインフォマティクスの解析をすでに終えて公共のデータベースに登録されている小さなサイズのデータを扱っています。それでもデータ量を増やすと、ダウンロードやコンピュータの実行時間が大変になっていきますし、やがてメモリを気にする必要も出てくるかもしれません。
そこで遺伝子の数を絞ることを考えます。ここではKEGGに胃癌関連と登録されている遺伝子に絞ることにします。
KEGGの GASTRIC CANCERのパスウェイの図
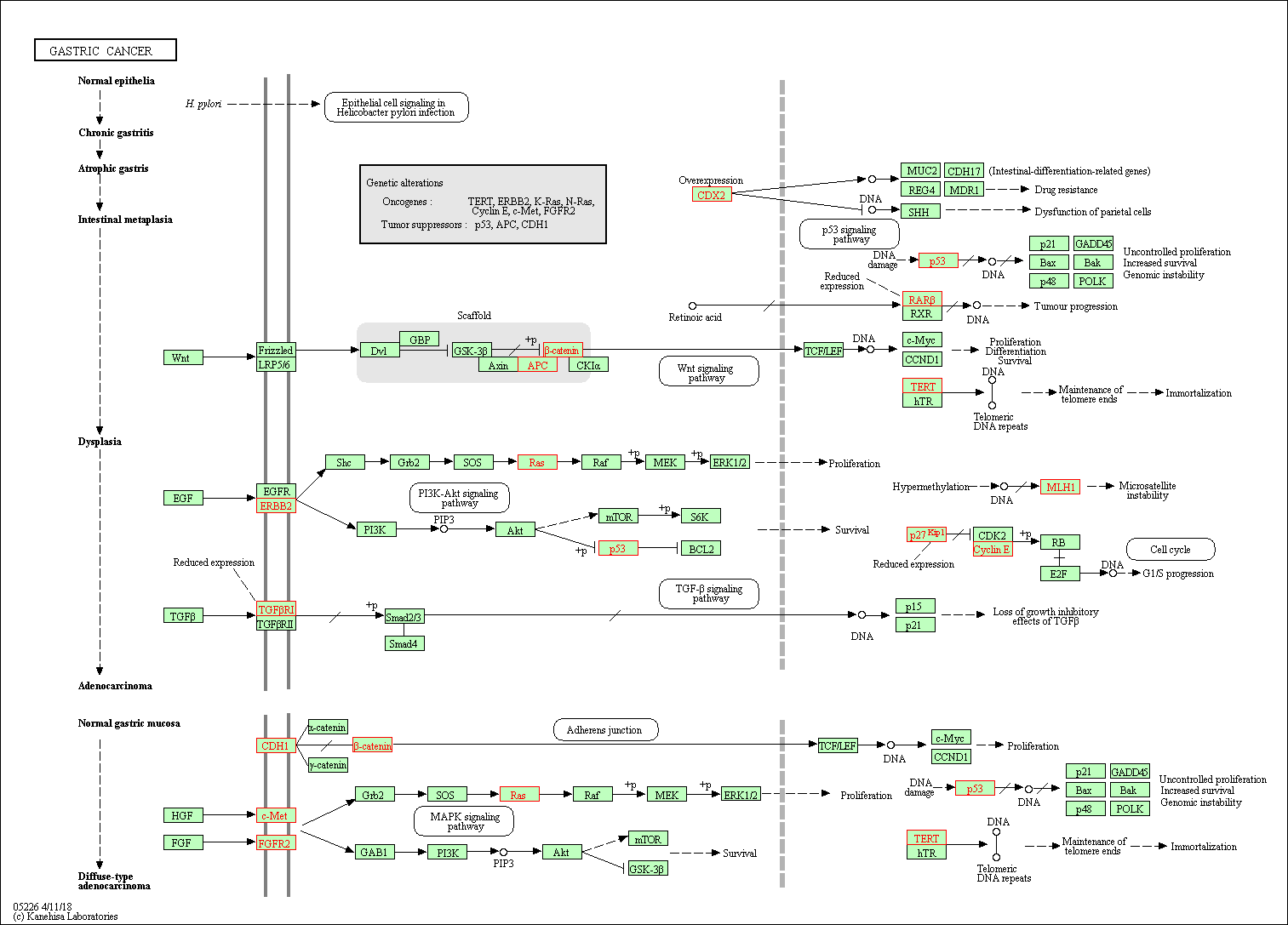
KEGG謹製のAPIに直接アクセスしてもよいのですが、ここではより簡単にTogoWSを使用します。
## TogoWS用にクライアントの作成
tgw = Daru::APIClient.new "http://togows.org"
## 取得
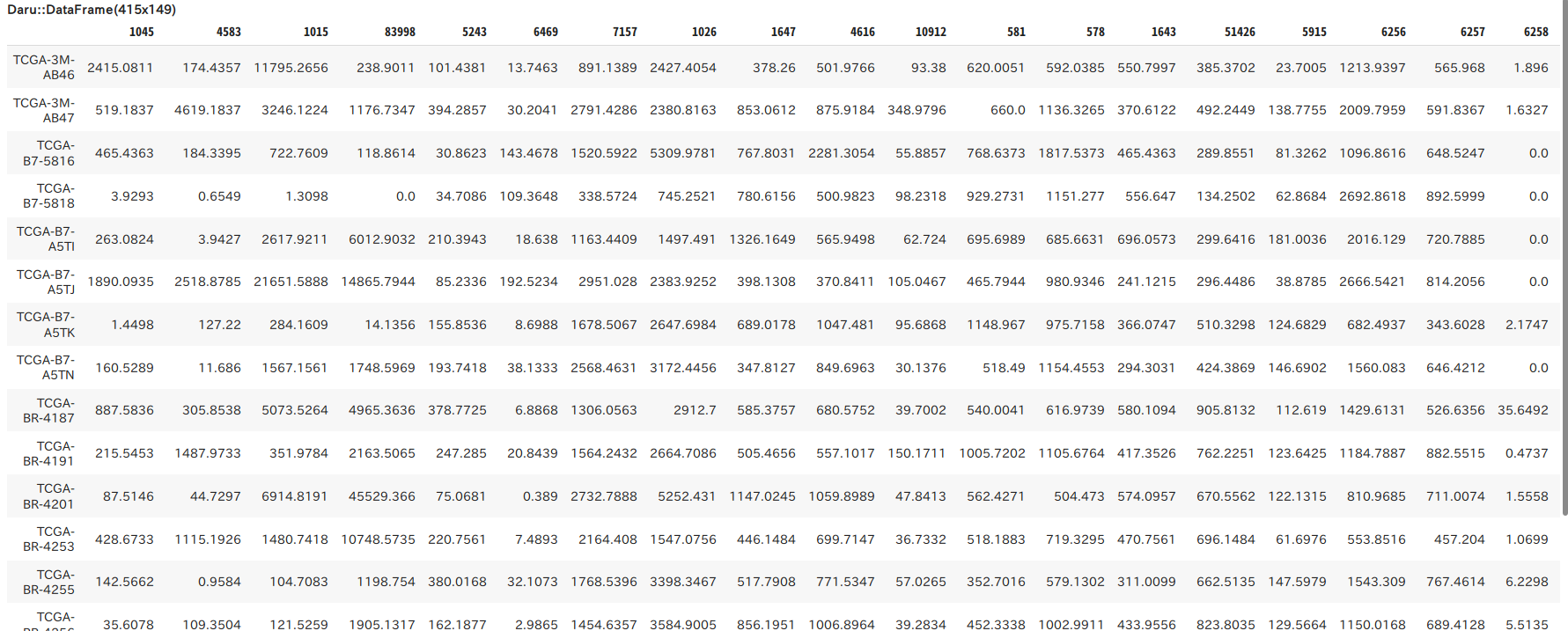
gastric_genes = tgw.get "/entry/kegg-pathway/hsa05226/genes.json"
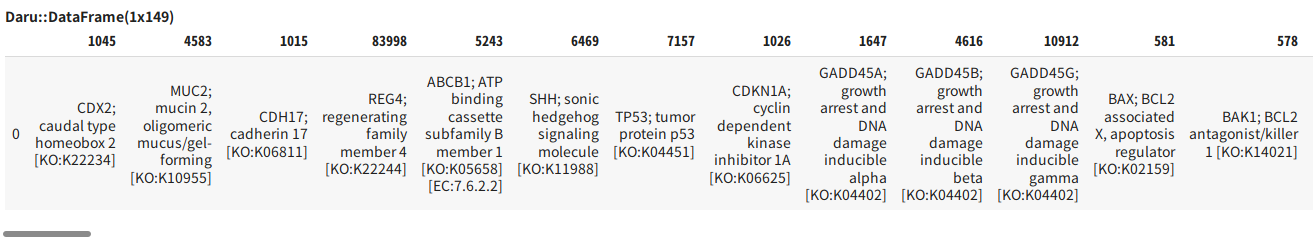
こんな感じで149の遺伝子がEntrezGeneIdとともに取得できれば成功です。
entrezGeneIdを配列にます。(配列だけでなくデータフレームもあとで使います)
# GeneID を Array に変換!
gastric_gene_ids = gastric_genes.vectors.to_a

EntrezGenIdを指定して、mRNAの発現量を取得する
cBioPortalからmRNAの発現量を取得します。
ここでは、get ではなく post で先ほど取得した胃癌の遺伝子のIdリストをサーバーに渡します。そのため少し記述量が多くなります。
if File.exists? "mrna_expressions.csv"
mrna_expressions = Daru::DataFrame.from_csv "mrna_expressions.csv"
else
mrna_expressions = c.post(
"/molecular-profiles/stad_tcga_rna_seq_v2_mrna/molecular-data/fetch",
headers: {"Content-Type": "application/json"},
body: {entrezGeneIds: gastric_gene_ids,
sampleListId: "stad_tcga_rna_seq_v2_mrna"}.to_json)
mrna_expressions.write_csv "mrna_expressions.csv"
end
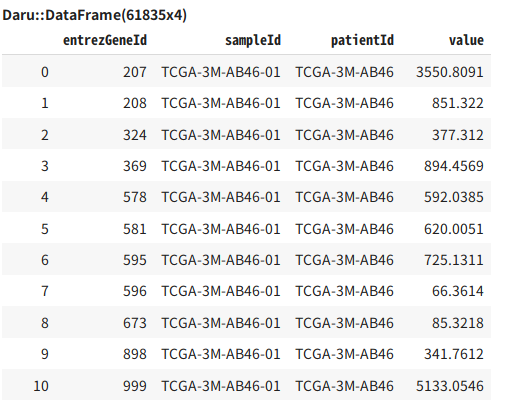
mrna_expressions["entrezGeneId", "sampleId", "patientId", "value"]
(毎回ではありませんが、この工程に時間がかかるケースがあるようです。なので、データを一回取得したらcsvファイルに保存するようにしてあります。)
下図のような結果が出力されれば成功です。
(省略)
サンプルIDごとに結果をまとめる
もっとよいやり方があると思われますが、私は以下のようにしました。
先程TogoWSから取得した胃がんの遺伝子リストのデータフレームを流用します。
samples = mrna_expressions.group_by("sampleId").each_group.to_a
result = gastric_genes.dup
samples.each do |df|
df2 = df.only_numerics
df2["entrezGeneId"].map!{|i| i.to_s} # stringにしないとマッチしない
df2.set_index "entrezGeneId"
result.row[df.patientId[0]] = df2["value"]
end
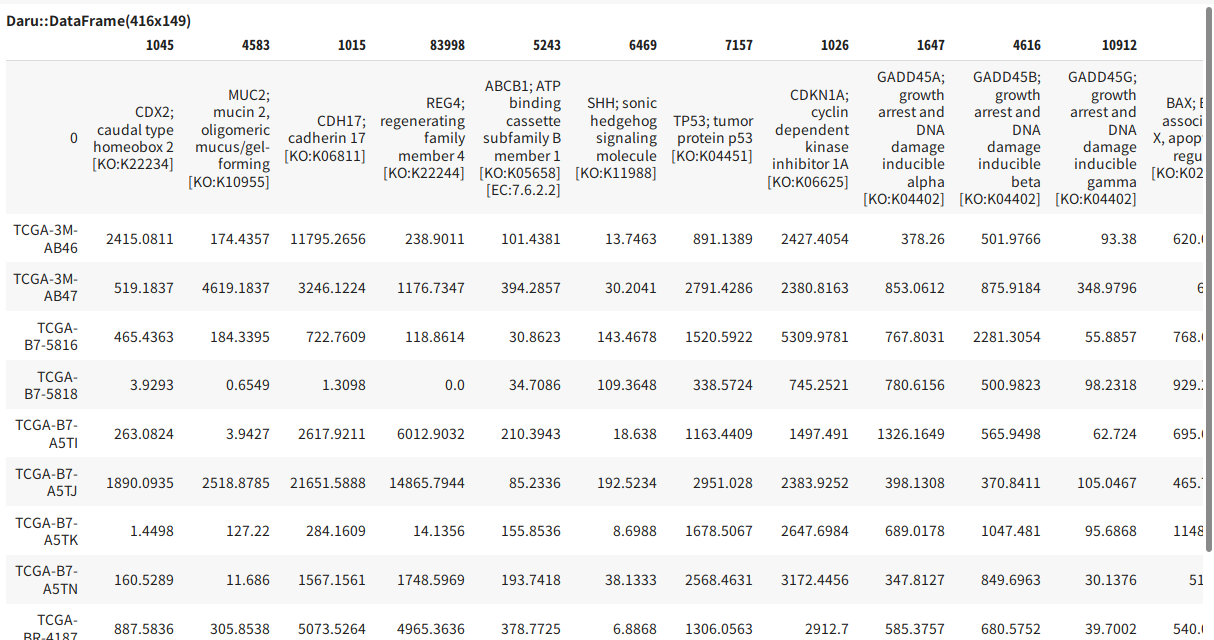
result.head
(この時点では1行目が残っています)
(Zスコアを選択した場合は、ここで空欄が発生するので注意してください)
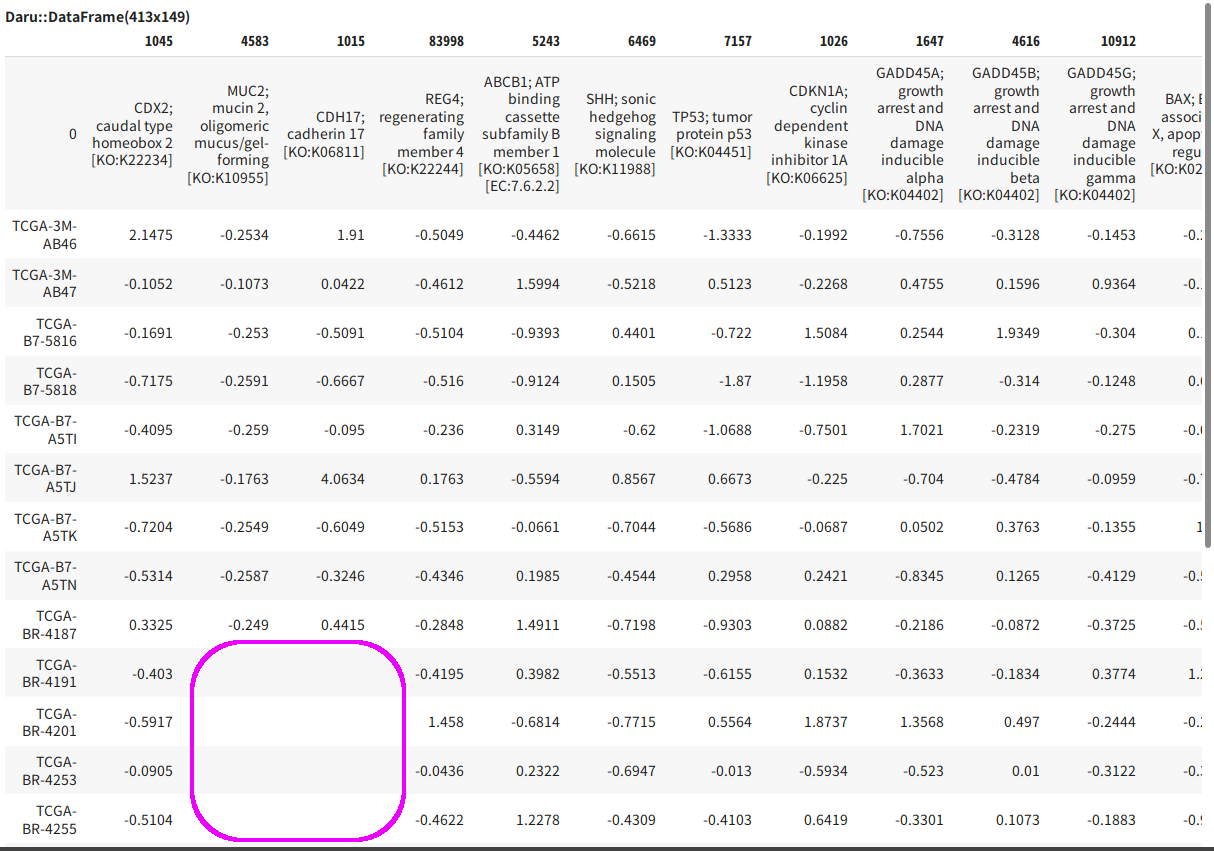
欠損値がないかは、次のような感じで調べることができます。
(なぜここだけcollectかというと、Daruではmapとcollectが異なる挙動をするからです![]() )
)
result.collect{|v| v.count_values nil}
TCGAで取得できる臨床データ
本筋から外れますが、このスタディでどのような臨床データが取得できるのか見ておきます。
c.get("/studies/stad_tcga/clinical-attributes")
3 分で作る無料の翻訳 API with Google Apps Scriptを利用して項目名だけ日本語に翻訳してみました。
こんな感じです
[
[ 0] "年齢",
[ 1] "AJCC転移病理PM",
[ 2] "AJCC病棟病理学PN",
[ 3] "AJCC病理腫瘍病期",
[ 4] "AJCCステージングエディション",
[ 5] "AJCC腫瘍病理学PT",
[ 6] "バレット食道",
[ 7] "がんの種類",
[ 8] "がんの種類",
[ 9] "調達都市",
[10] "臨床病期",
[11] "CLIN M STAGE",
[12] "クリンステージ",
[13] "CLIN T STAGE",
[14] "誕生日",
[15] "収集する日数",
[16] "死ぬまでの日数",
[17] "初期の病理診断までの日数",
[18] "最後のフォローアップの日数",
[19] "患者への無増悪期間",
[20] "コレクションの日数",
[21] "腫瘍進行までの日数",
[22] "DFS月",
[23] "DFSの状況",
[24] "疾患コード",
[25] "人種",
[26] "外節への関与",
[27] "胃癌の家族歴",
[28] "フォーム記入日",
[29] "フラクションゲノムの変更",
[30] "グレード",
[31] "組織学的診断",
[32] "歴史NEOADJUVANT TRTYN",
[33] "その他の悪性腫瘍",
[34] "Hピロリ感染症",
[35] "ICD 10",
[36] "ICD O 3の歴史",
[37] "ICD O 3 SITE",
[38] "検証済みの同意済みの確認",
[39] "初期病理DX年",
[40] "IS FFPE",
[41] "最長寸法",
[42] "調べたリンパ節",
[43] "リンパ節は彼が数えた調べた",
[44] "リンパ節検査数",
[45] "サンプル調達方法",
[46] "突然変異カウント",
[47] "初期治療後の新しい腫瘍イベント",
[48] "IHCで陽性のリンパ節数",
[49] "胃がんの関連数",
[50] "埋め込まれたOCT",
[51] "オンコツリーコード",
[52] "OSの月",
[53] "OSの状況",
[54] "サンプル調達の他の方法",
[55] "他の患者ID",
[56] "他のサンプルID",
[57] "病理学報告書ファイル名",
[58] "病理報告書UUID",
[59] "患者の死亡原因",
[60] "一次サイトの患者",
[61] "プロジェクトコード",
[62] "見込みコレクション",
[63] "レース",
[64] "放射線治療アジュバント",
[65] "REFLUXの歴史",
[66] "残存腫瘍",
[67] "回顧コレクション",
[68] "サンプル数",
[69] "サンプル初期重量",
[70] "サンプルの種類",
[71] "サンプルタイプID",
[72] "SEX",
[73] "最短寸法",
[74] "腫瘍組織の部位",
[75] "検体の現在の重量",
[76] "検体凍結方法",
[77] "2番目に長い最長寸法",
[78] "他のステージ",
[79] "対象となる分子治療",
[80] "クランピングとフリーズの間の時間",
[81] "強制終了と凍結の間の時間",
[82] "組織のソースサイト",
[83] "処理",
[84] "治療アウトカムファーストコース",
[85] "治療タイプ",
[86] "腫瘍サンプル採取国",
[87] "腫瘍の状態",
[88] "バイアル番号",
[89] "バイタルステータス"
]
コメント = <<'EOS'
自動翻訳がおかしいところがありますが、全体の傾向はわかると思います。
登録されている臨床データは、スタディーによって違います。
データの収集には、専門職の判断や、入力作業など人間が必ず介在します。
人間が介在している以上、ごく少量のミスは必ず混じっていると考えた方が自然です。
たとえ登録の段階で一つもミスが発生しないとしても
本人が自分の情報、たとえば誕生日や人種を誤って申告してしまうようなケースも、
サンプル数が十分に大きいときは十分に発生してしまいます。
いずれにしろ、あらゆるレイヤーでミスは発生しますので、
データセットの情報を過度に盲信すべきではないと思います。
これは臨床情報だけではなく、遺伝子情報についてもおそらく同様です。
検体の取扱には、必ず人手が介入するシーンがあります。
人間が介在している以上、ごく少量のミスは必ず混じると考えた方が自然です。
スタディーによって、検出精度には大きな違いを認め、
中には不十分な処理を行っているものもあるかもしれません。
そうでなく全て適切な処理を行っていたとしても、
使用した検査機器・パネル・使用したリファレンスゲノムのバージョンなどによって、
かなり異なる結果が出ている可能性がありそうです。
しかし、これを考えているとキリがなくなるので先に進みます。
EOS
リンパ節転移の情報を取得する
TNM分類の情報が登録されているのでこれを利用します。
T(tumor)
腫瘍(原発巣)の大きさと進展度を表す。T1〜4までの4段階に分けられる。N(nodes)
所属リンパ節への転移状況を表す。転移のないものをN0とし、第一次リンパ節、第二次リンパ節への転移、周囲への浸潤の有無から N3までの段階に分ける。M(metastasis)
遠隔転移の有無を表す。遠隔転移がなければM0、あればM1となる。
この分類でN0なら、リンパ節転移なし、N1以上なら転移ありということになります。
この情報はclinical-dataから取得できます。
ln = c.get "/studies/stad_tcga/clinical-data",
query: {clinicalDataType: "PATIENT", attributeId: "AJCC_NODES_PATHOLOGIC_PN"}
ln.at 1..4
(clinicalDataType には Patient と Sample の2種類があります。これは一人の患者さんから複数の検体が採取されているケースがあり、一対一対応しないためです)
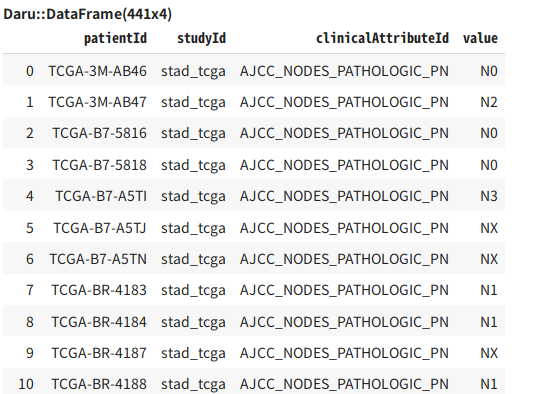
先ほど書いた通り、N0は転移なし、N1以上は転移ありの意味です。NXは「不明」の意味だと思いますので、NXのサンプルは除外する必要があります。それぞれいくつあるのか数えてみます。
ln.value.value_counts

データフレームのマージ機能などを使用した方がよいのかもしれませんが、簡単なのでRubyのHashをつかって、patientIDからリンパ節転移の記号を返す辞書を作成してしまいましょう。keyにpatientId valueにvalueを選択しています。
keys = ln["patientId"].to_a
values = ln["value"].to_a
lnh = keys.zip(values).to_h
NX(不明) の検体を除きます
nx_patient_ids = ln.where(ln["value"].eq "NX").patientId.to_a
nx_patient_ids.each do |id|
result.delete_row id
end
以上で、必要なデータをcBioPortalで準備し終わりました。
Rumaleを使用した機械学習
ここからは、Rubyでも機械学習ができると評判のRumaleというライブラリを使用します。
Rumaleの作者のブログの下記のページを参考にコピー&ペーストしながら実行していきます。
ラベルを作成する
labels = result.index.to_a.map do |vn|
case lnh[vn]
when "N0" # リンパ節転移なし
0
else # それ以外
1
end
end
# Rumale で使用するために、Int32に変更します
labels = Numo::Int32[*labels]
サンプルを作成する
samples = Numo::DFloat[*result.to_matrix.to_a]
p samples.shape # [398, 149]
(注:2019/04/18現在、JupyterのRubyカーネルであるIRubyは大きなサイズのArrayを表示するのに向いていない)
モデルを作成する
ランダムフォレストを使用します。
model = Rumale::Ensemble::RandomForestClassifier.new(random_seed: 1)
交差検定
ここはすべて公式ブログからのコピペです。実はあまりわかっていませんが、ありがたく使わせてもらいます。
# 結果を保存する変数を初期化する
sample_n = result.vectors.to_a.size
imp = Numo::DFloat.zeros(sample_n)
sum_accuracy = 0.0
# 10-交差検定の分割をおこなう
kf = Rumale::ModelSelection::StratifiedKFold.new(n_splits: 10, shuffle: true, random_seed: 1)
kf.split(samples, labels).each do |train_ids, valid_ids|
# 訓練データと検証データを得る
train_samples = samples[train_ids, true]
train_labels = labels[train_ids]
valid_samples = samples[valid_ids, true]
valid_labels = labels[valid_ids]
# Random Forestで学習する
clf = Rumale::Ensemble::RandomForestClassifier.new(n_estimators: 100, max_features: 2, random_seed: 1)
clf.fit(train_samples, train_labels)
# 特徴量の重要度を得る
imp += clf.feature_importances
# 正確度を得る
sum_accuracy += clf.score(valid_samples, valid_labels)
end
0
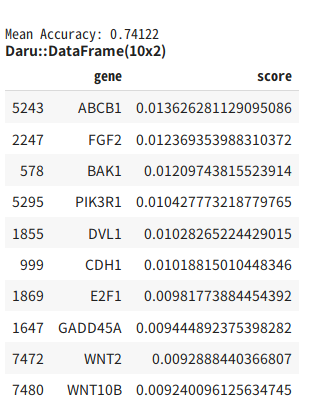
結果の表示
# 平均の正確度を出力する
mean_accuracy = sum_accuracy / kf.n_splits
puts
puts sprintf("Mean Accuracy: %.5f", mean_accuracy)
# 特徴量の重要度を出力する
raise if result.vectors.to_a != gastric_gene_ids # 念の為確認
r = Daru::DataFrame.new({
gene: gastric_genes.row_at(0).map{|i| i.split(";")[0]},
score: imp},
index: gastric_gene_ids
).sort([:score], ascending: false) / kf.n_splits
r.head(10)
結果は以下のようになりました
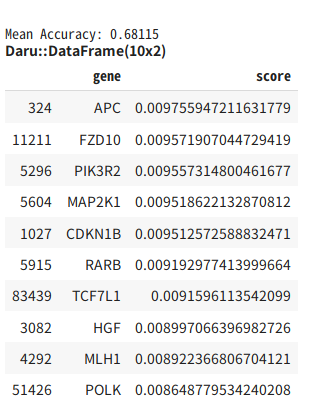
予想の正確度は68%で、偶然よりかなり高い確率で予想できていると思われますが、ものすごく高い数字かというと微妙です。また、これらの遺伝子は、リンパ節への転移のしやすさというよりも、単に進行の度合いを表している可能性も高いでしょう。
そこで、上記のコードを書き換えて T1・T2 / T3以上 に関しても同じことをやってみました。(コード省略します)
予想精度はリンパ節の時よりもかなりよさそうです。
それでは、T分類における重要度の高さをx軸、N分類における重要度の高さをy軸として、Daru::Viewを使って散布図にしてみます。
Daru::View::Plot.new(r3,
type: :bubble,
hAxis: {title: "T12/34"},
vAxis: {title: "N0/その他"},
sizeAxis: {maxSize: 20},
height: 1000,
width: 1000,
bubble: {textStyle: {fontSize: 11}}
).show_in_iruby```
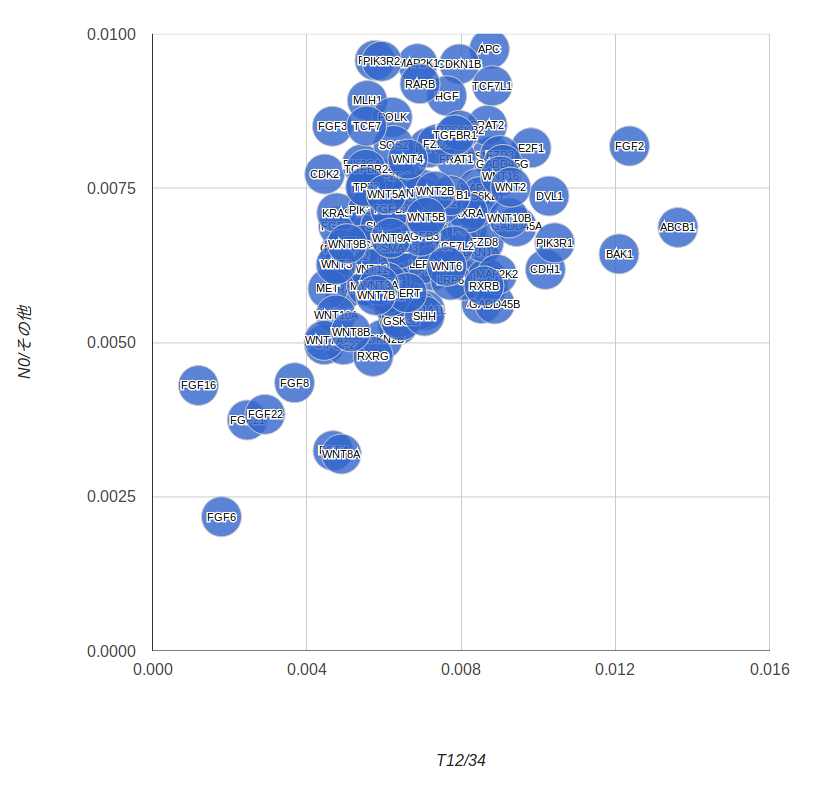
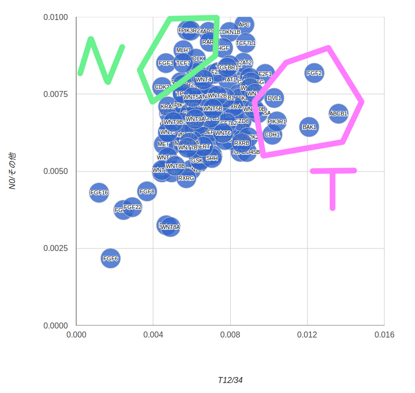
このグラフを見ると、FZD10, PIK3R2, MLH1, FGF3, CDK2
あたりは、浸潤よりもリンパ節転移の方に強く関係しているように見えます。本当かなあ…。
インターネットを軽くググっても、これらの遺伝子がリンパ節転移に関係あるというような話はあまり出てこないので、正直この結果はあやしいのです。
終わりに
cBioPortalの公開しているゲノムのデータをRubyで機械学習してみました。ゲノムのデータは、サンプル数に比較して特徴量が膨大で、何らかの知見を使用して特徴量を強力に絞り込まなければならないと感じました。これが、いわゆる世間で言われている次元の呪いというやつでしょうか。
また、一番最初のところで、「mRNAの発現量はリンパ節転移を予想できるか」という目標を設定しましたが、コンピュータを使った解析は、特定の仮説を検証するよりも、網羅的に生成した仮説を調べてあとで人間の感覚と比較するやり方の方が相性がよいかもしれないと思いました。上の検討を継続するなら、N01/N23 も調べて M(遠隔転移)も見てみたいところです。
正しく実行できているとはあまり思っていなくて、多分…いや絶対ミスとかあると思います。しかしRuby会議中に公開しようと思いました。もしも、なにか気がついたことがあったらぜひコメントして教えてもらえると嬉しいです。
この記事は以上です。