小ネタです。
アウトプットしないと何も覚えないので書いてます。R初心者のため低クオリティです。
TCGAの論文とかに出てくる、がんのゲノム変異の一覧みたいな図をOncoPlotと言うそうです。
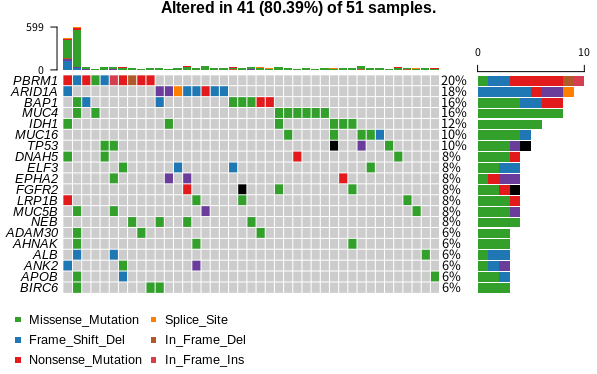
これを作ってくれるRのパッケージがmaftoolsです。bioconductorからインストールします。
https://bioconductor.org/packages/release/bioc/html/maftools.html
maftoolsは、mafファイルを必要とします。VCFファイルから、vcf2mafという使いにくそうなツールを使って生成するようですが、TCGAのサイトからmafファイルをダウンロードする方法もあります。そのためのパッケージがTCGAbiolinksです。
それでは、TCGAbiolinksのチュートリアルと全く同じネタですが、胆管がんのOncoplotを作成してみす。
library(TCGAbiolinks)
library(maftools)
library(dplyr)
maf <- GDCquery_Maf("CHOL", pipelines = "varscan2") %>% read.maf
Rユーザーにとっては常識の類だと思われますが、%>% はmagrittrの提供する機能だそうです。よくわかりませんがコマンドラインの|に似ている機能だと想像します。
mode(maf)
とやると、[1] "S4"と表示されます。このmafの中味はS4クラスというやつなのだろうと思われます。
脱線しましたが、oncoplotを描きます。
oncoplot(maf = maf)
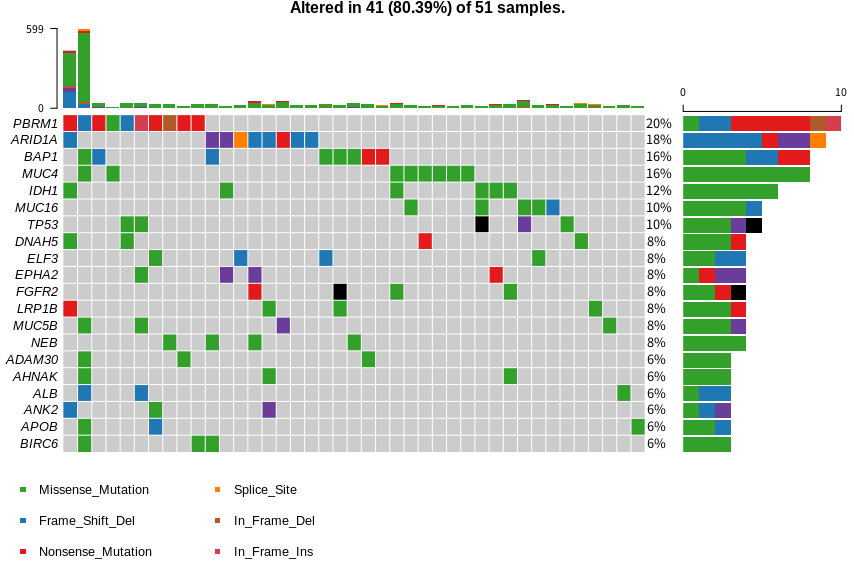
簡単ですね。
臨床のデータと組み合わせるときは、
library(TCGAbiolinks)
library(maftools)
tcgacode = "CHOL"
barcode2simple <- function(x) {
ar = strsplit(x , "-")
paste(ar[[1]][1:3], collapse = "-")
}
maf = GDCquery_Maf(tcgacode, pipelines = "varscan2")
maf["Tumor_Sample_Barcode"] = apply(maf["Tumor_Sample_Barcode"], 1, barcode2simple)
clinicalData = GDCquery_clinic(project = paste("TCGA", tcgacode, sep="-"), type='clinical')
names(clinicalData)[names(clinicalData)=="submitter_id"] = "Tumor_Sample_Barcode"
clinicalData["Tumor_Sample_Barcode"] = apply(clinicalData["Tumor_Sample_Barcode"], 1, barcode2simple)
head(clinicalData)
maf = read.maf(maf = maf, clinicalData = clinicalData)
oncoplot(maf = maf,
clinicalFeatures = c('gender','tumor_stage'),
additionalFeature = c("Tumor_Seq_Allele2", "C"),
draw_titv = TRUE,
top = 30)
みたいな感じにするとできるようです。(Rの初心者なので、上のコードは何やら非効率なことをやっている可能性が高いです
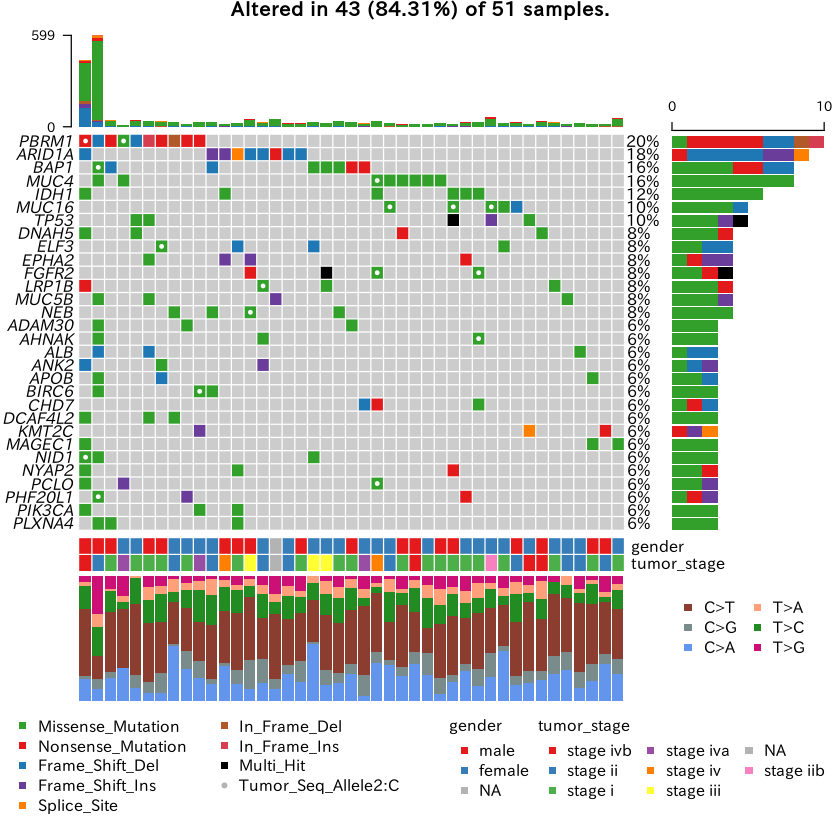
この記事は以上です。