はじめに
IGVはゲノムブラウザと呼ばれるソフトウェアです。アライメントされたリードのbamファイルや、ゲノム上の位置情報にアノテーションを加えたbedファイル、遺伝子情報の書かれたgff3ファイルなどさまざまなファイルを閲覧することができます。bioinformaticsに関わったことがある方は、このソフトをご存知の方が多いのではないでしょうか。
GithubのIGVのリポジトリ

※画像はIGVのホームページより
そんなIGVですが、マウスでクリックしながら操作すると少し手間がかかることがあります。
- リファレンスゲノムを指定して
- 必要なファイルを読み込んで
- 注目している場所を一つずつみてまわって
- スクリーンショットをとって画像として保存する
目視で結果を確認することは楽しいことでもあり、とても大事な作業だと思いますが、一方で定型的な作業はなるべく自動化していきたいという気持ちもあると思います。そこでIGVの操作をプログラミング言語から行うことを考えます。
※ 最近はJavaScriptで作られたigv.jsというものが活発に開発されて広く使われるようになっているようです。これに関しては調査が不十分なのでこの記事では触れません。(しかしいずれはQiita記事にしてきたいと思います。)
ソケット通信を利用してIGVを操作する
実はIGVはソケット通信を利用してport 60151番から操作できるようになっています。
メニューバーの View > Preference の中に advanced というタブがあります。Enable portにチェックが付いていない時は、ここをチェックします。
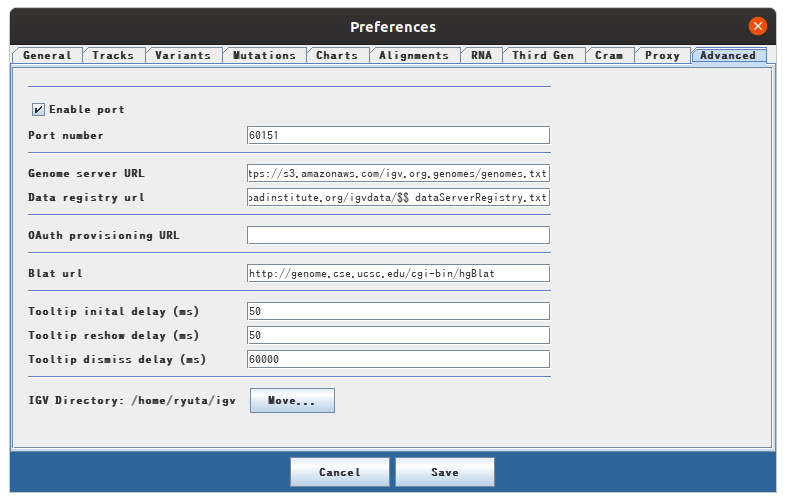
するとport 60151番を使って、ソケット通信を利用してIGVを操縦することができるようになります。
IGVの操作に使えるコマンドの一覧
公式リファレンスをDeepL翻訳を使いながら日本語に翻訳してみました。
| Command | Description |
|---|---|
| new | 新しいセッションを作成します。 デフォルトのゲノムアノテーション以外のすべてのトラックを取り除きます。 |
| load file | データまたはセッションファイルを読み込みます。フルパスまたはURLをカンマ区切りで指定します。 |
| collapse trackName | 指定されたtrackNameを折りたたむ。trackNameを指定しない場合はすべてのトラックが折りたたまれます。 |
| echo | レスポンスに "echo "を返します。 (テスト用) |
| exit | IGVアプリケーションを終了します。 |
| expand trackName | 指定したtrackNameを展開します。trackNameを指定しない場合はすべてのトラックが展開されます。 |
| genome genomeIdOrPath | idでゲノムを選択、または指定されたパスからゲノム(インデックス化されたfasta)をロードします。 |
| goto locus or listOfLoci | 単一の遺伝子座またはスペースで区切られた遺伝子座のリストにスクロールします。リストが提供されている場合は、これらの座位が分割スクリーンビューで表示されます。 IGV検索ボックスで有効な構文ならどれでも大丈夫です。 |
| goto all | ゲノム全体の表示にスクロールします。 |
| group option | アライメントトラックのみ。 次のオプションのいずれかでアライメントをグループ化します。 STRAND, SAMPLE, READ_GROUP, LIBRARY, FIRST_OF_PAIR_STRAND, TAG, PAIR_ORIENTATION, MATE_CHROMOSOME, SUPPLEMENTARY, MOVIE, ZMW, HAPLOTYPE, READ_ORDER, NONE, BASE_AT_POS |
| region chr start end | 2つの遺伝子座で囲まれた関心領域を定義します(例えば、region chr1 100 200)。 |
| maxPanelHeight height | 画像に含める各パネルの縦方向のピクセル数(高さ)を設定します。ポートコマンドまたはバッチスクリプトから作成された画像は、画面上に表示されるデータに限定されません。別の言い方をすれば、スクロール可能な画面領域に表示されている部分だけでなく、パネル全体を画像に含めることができます。この設定のデフォルト値は1000で、より多くのデータを表示するにはこの値を大きくし、より小さい画像を作成するにはこの値を小さくします。 |
| setLogScale(true or false) | |
| setSleepInterval ms | 遅延(スリープ)時間をミリ秒単位で設定します。 スリープ間隔は、連続するコマンド間に呼び出されます。 |
| snapshotDirectory path | 画像を書き込むディレクトリを設定します。 |
| snapshot filename | IGV ウィンドウのスナップショットを画像ファイルに保存します。 filenameを省略した場合、軌跡に基づいて生成されたファイル名を持つPNGファイルを書き込みます。 filenameが指定された場合、ファイル名の拡張子によって画像ファイルの形式が決定され、.png、.jpg、または.svgでなければなりません。 |
| sort option locus | アライメントまたはセグメント化されたコピーナンバーのトラックをソートします。 セグメント化されたコピーナンバーのoptionに適応される値は、(1)セグメント化されたコピー番号のAMPLIFICATION と DELETION、(2)アライメントトラックのPOSITION、STRAND、BASE、QUALITY、SAMPLE、READGROUP、INSERSTSIZE、FIRSTOFPAIRSTRAND、MATECHR、READORDER、およびREADNAMEです。 optionは、大文字と小文字を区別しません。 locusを指定すると、単一の位置または範囲を定義することができます。 オプションを指定しない場合は、表示されている領域、または表示されている領域の中心位置に基づいてソートが実行されます。 |
| squish trackName | 与えられた trackName を Squish します。 trackName はオプションで、指定しない場合はすべてのアノテーショントラックが Squish されます。 |
| viewaspairs trackName | アライメントトラックの表示モードを "View as pairs "に設定します。 |
| preference key value | keyという名前の設定を指定した値に一時的に設定します。この設定は、IGVがシャットダウンされるまでのみ有効です。 |
IGVをプログラミング言語から操る
Java
IGVはJavaで開発されているソフトウェアなので、公式のexampleもJavaで書かれています。
Socket socket = new Socket("127.0.0.1", 60151);
PrintWriter out = new PrintWriter(socket.getOutputStream(), true);
BufferedReader in = new BufferedReader(new InputStreamReader(socket.getInputStream()));
out.println("load na12788.bam,n12788.tdf");
String response = in.readLine();
System.out.println(response);
out.println("genome hg18");
response = in.readLine();
System.out.println(response);
out.println("goto chr1:65,827,301");
//out.println("goto chr1:65,839,697");
response = in.readLine();
System.out.println(response);
out.println("snapshotDirectory /screenshots");
response = in.readLine();
System.out.println(response);
out.println("snapshot");
response = in.readLine();
System.out.println(response);
R
さて、Java以外のプログラミング言語ではどうでしょうか。Rを使っている場合は、あまり詳しくないですが、bioconductorあたりからIGVを操作するライブラリが提供されているでしょうから、それを使えばよいでしょう。少し検索するだけでも igvRというソフトウェアがヒットします。これは igv.jsを活用するものらしいので、デスクトップのIGVを自動操作するものではないかも知れませんが…。
Python
少し古いスクリプトですが、最近精力的にNim言語を使用してバイオインフォマティックスのツールを開発されているBrent Pedersenさんが作成した igv.pyというツールがあります。これは上記のソケット通信をラップした小さなライブラリです。
Ruby
私はRubyが好きです。ナニナニ? ツールが少ない? それなら自分で作ってしまえばいいでしょうが!
ということで、上記のBrent Pedersenさんのスクリプトをまるっと参考にして、Ruby言語からIGVを操作できる ruby-igv というツールを作成しました。これでRuby言語からも簡単にIGVを操作できるようになります。
使い方はこんな感じです。
igv = IGV.new
igv.load 'na12788.bam'
igv.genome 'hg18'
igv.go 'goto chr1:65,827,301'
igv.save 'image.png'
まだ出来たてホヤホヤのツールです。
粗削りのところとか、バグとかに遭遇した場合、あるいは要望を見つけましたらぜひGithubのissueに報告してください。もちろんプルリクエストも歓迎します。
おわりに
bioinformaticsに限らずですが、どうしても、どうすれば既存の道具を組み合わせて目的が達成できるか、どうすればツールを使いこなせるかという点に注目が集まりがちです。そして、せこせこと道具を作っていると、目的と手段が逆転しているなどという風にとらえられることもありえます。
でもそれは、ある意味では、とてもセルフィッシュな視野の狭い考え方だと思うんですよね。もっと道具を作って公開する人がいれば、それだけ世界は便利になって広がって豊かになっていきます。みんなもっと自由に道具を作って公開しよう。(別にRubyじゃなくてもいいので)