SCNICとは
SCNICとは、"Sparse Cooccurrence Network Investigation for Compositional data"の略であり、「組成データのための疎な共起ネットワーク分析」のことで、SparCCという相関係数の計算アプローチを用いて、マイクロバイオームの組成データの相関関係を推定するツール。
ここでいう組成データというのは、DADA2などで作成されたASVのテーブルデータや、SILVAデータベースなどにアサインして得た系統組成データを指す。
今回は、QIIME2にモジュールとして組み込まれている共起ネットワーク分析ツールq2-SCNICの使い方を説明する。
相対量データにおける相関関係推定の難しさ
16S rRNAアンプリコンシーケンスなどでマイクロバイオームの解析をするとき、グループ間の微生物コミュニティの相関関係を推定することが多い。
しかし、次世代シーケンサによって得られたマイクロバイオームのデータを相対量に変換して解析を進めざるを得ないため、いろいろと解釈が難しくなってしまう。
QIIME2でいうと バラついたサンプルごとのリード数をサブサンプリングで正規化する「希薄化」 がこれにあたる。
(16Sコピー数で正規化する方法もあるが、すべての細菌においてコピー数が知られているわけではないので、推奨されないことが多い。)
このように相対量に変換してしまうと、 ある細菌のASV(またはOTU)が1つ増加すると、それ以外の細菌のASVが減少する必要があり、 本来は独立したASV同士であってもあたかも負の相関関係があるように見えてしまう 。
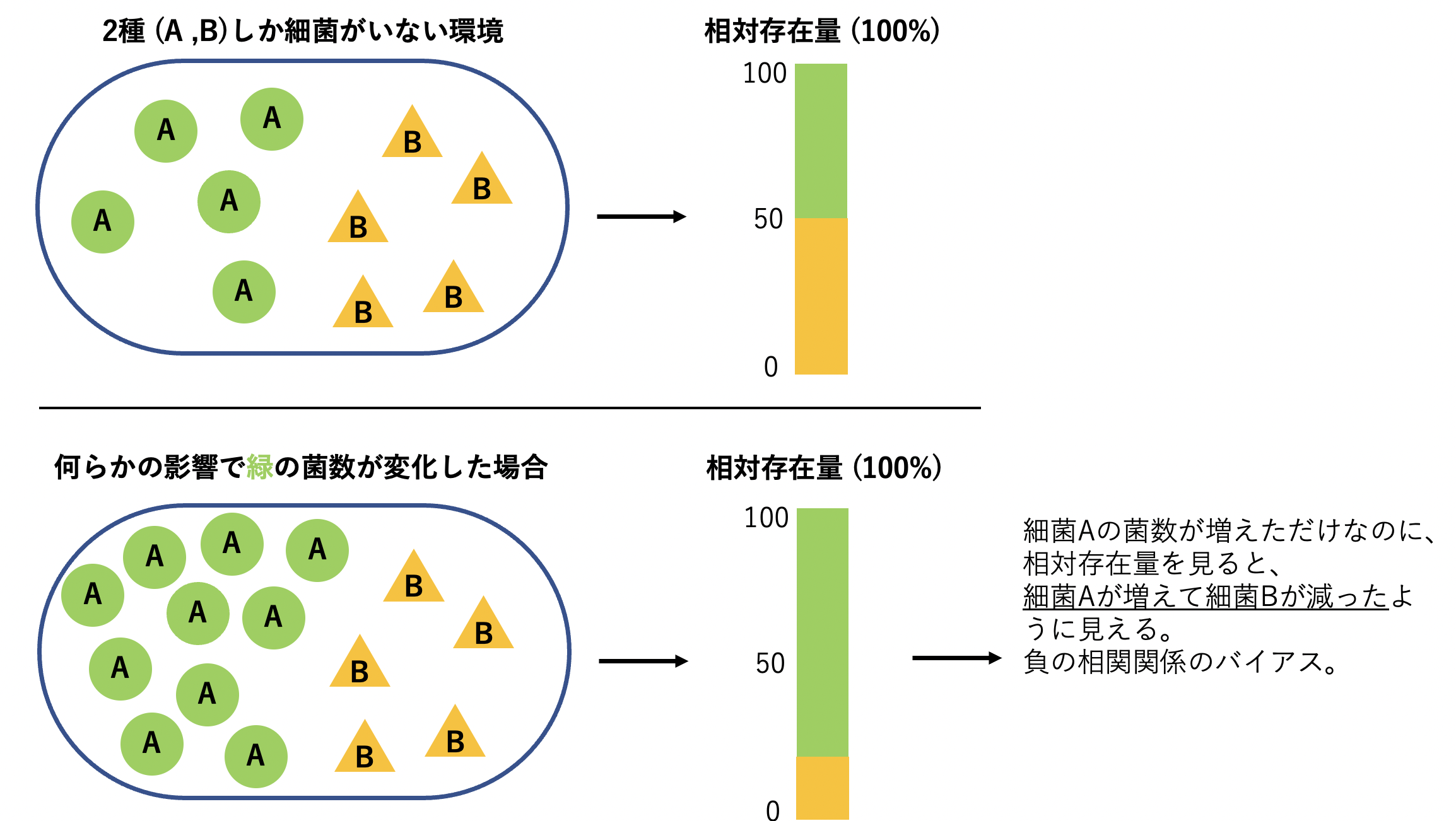
このような相対量のデータの相関関係を調べること自体が誤解を招くものだと言われている。
身体部位のマイクロバイオームデータをピアソン相関で推定し作ったネットワーク (左列)に対して、そのマイクロバイオームデータの相対存在量データをランダムにシャッフルしてピアソン相関で推定し作ったネットワーク(真ん中の列)は、なんと同様のパターンを示している。
ピアソン相関で得られたネットワークは、生物学的な特徴ではなく、相対量の構造的なバイアスに起因している ことがわかる。
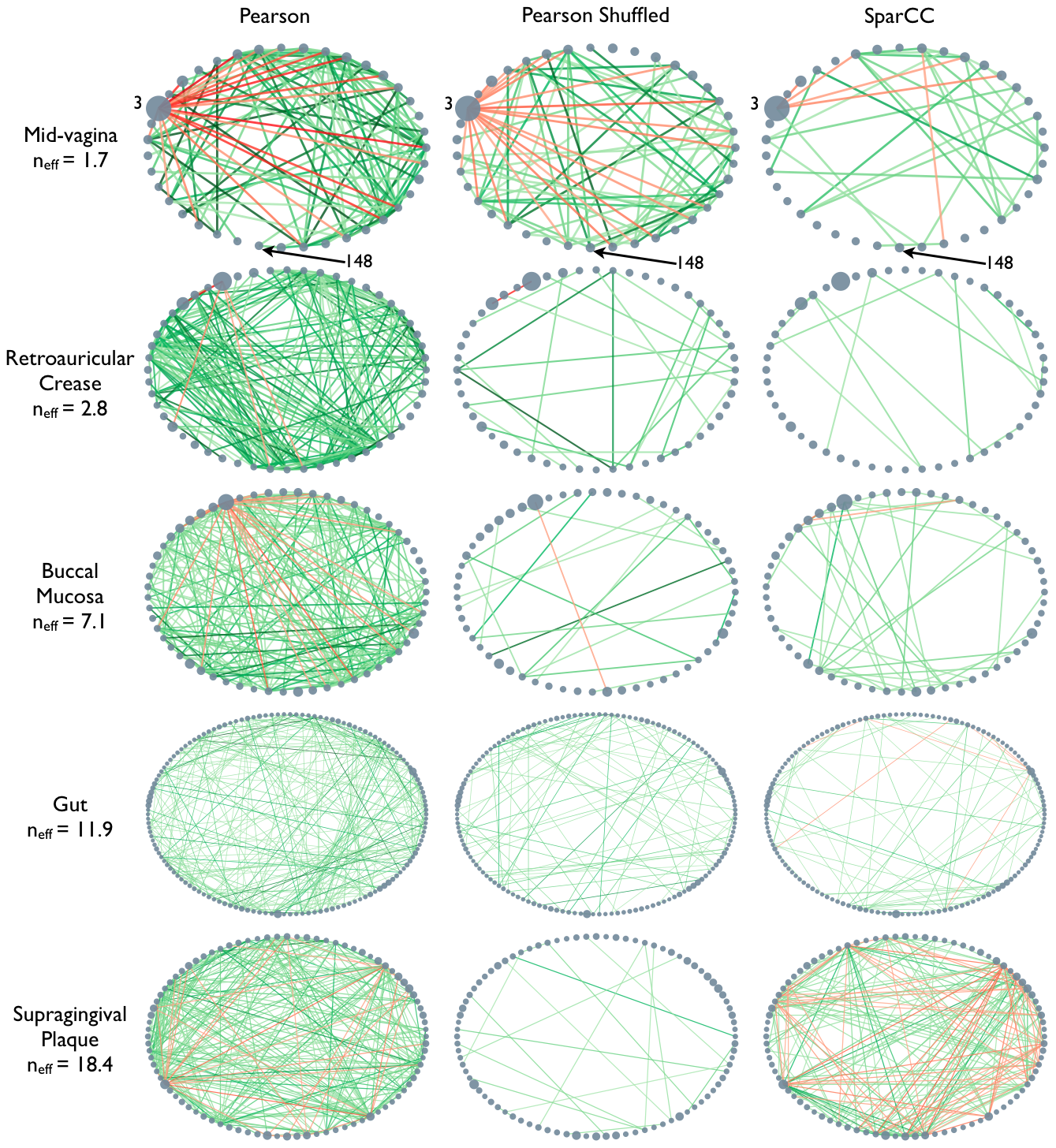
SparCCの論文より
Figure 1. Similar correlation networks are observed for real world vs. randomly shuffled bacterial abundance data.
SCNICでは、相関係数の算出にSparCCというアプローチの使用することで、希薄化(正規化)によって相対量に変換せずに、マイクロバイオームの組成データを用いてネットワーク分析が実施できる。
SparCCを用いた相関係数の計算
SparCCでは、
- 異なるASVの数が多い (= サンプル内の多様性が高い)
- 真の相関ネットワークは「疎」 (ほとんどのASVはお互いに強い相関はない)
と仮定して、対数変換したASV間の分散を用いて真のASVの分散を概算している。
ちなみに疎ではない組成データ(多くのASV間で強い相関が見られる)においても、かなりロバストだと書かれている。
q2-SCNICのインストール
condaでもpipでもインストール可能。
# QIIME2の起動(今回は2021.2バージョンで実行した)
conda activate qiime2-2021.2
# SCNICのインストール
conda install -q scnic
# プラグインのインストール
pip install git+https://github.com/shafferm/q2-SCNIC.git
# QIIME2のリフレッシュ
qiime dev refresh-cache
# 「fastsparがないよ」とエラーを出す場合はインストールする
conda install -c bioconda -c conda-forge --override-channels fastspar
解析の実行
0. テストデータの取得
テストデータとして、QIIME2 2021.4のドキュメントにある、おなじみの“Moving Pictures” tutorialからSequence quality control and feature table constructionのtable.qza をダウンロードする。
各サンプルの組成データを相対化しなくていいので、希薄化される前のASV数がバラついているtable.qza をすぐに使うことができる。
# データのダウンロード
wget https://docs.qiime2.org/2021.4/data/tutorials/moving-pictures/table.qza
1. 組成データのフィルタリング
組成データ内に大量に0があると相関分析が面倒になるので、「ASV数が500未満のサンプル」と「すべてのサンプルにおける平均存在量が2未満のASV」をすべて削除する。
qiime SCNIC sparcc-filter \
--i-table table.qza \
--o-table-filtered table_filtered.qza
2. 相関係数の算出とネットワーク生成
フィルタリングされた組成データのASV間のすべてのペアワイズの組み合わせで相関係数を算出する。
ここで、SparCCアプローチを指定する。
qiime SCNIC calculate-correlations \
--i-table table_filtered.qza \
--p-method sparcc \
--o-correlation-table correls.qza
3. モジュールの検出とサマリー作成
モジュールとは、高い相関が見られたASV間のネットワークのことで、これら相関関係ネットワークをそれぞれモジュール名をつけてクラスタリングする。
qiime SCNIC make-modules-on-correlations \
--i-correlation-table correls.qza \
--i-feature-table table.qza \
--p-min-r .35 \
--o-collapsed-table collapsed.qza \
--o-correlation-network net.modules.qza \
--o-module-membership membership.qza
公式ドキュメントを見ると、make-modules-on-correlation-tableのままだが、SCNICの最新バージョンではmake-modules-on-correlationsに変更されているので注意する。
--p-min-rは、最小の相関係数を指定するオプション。
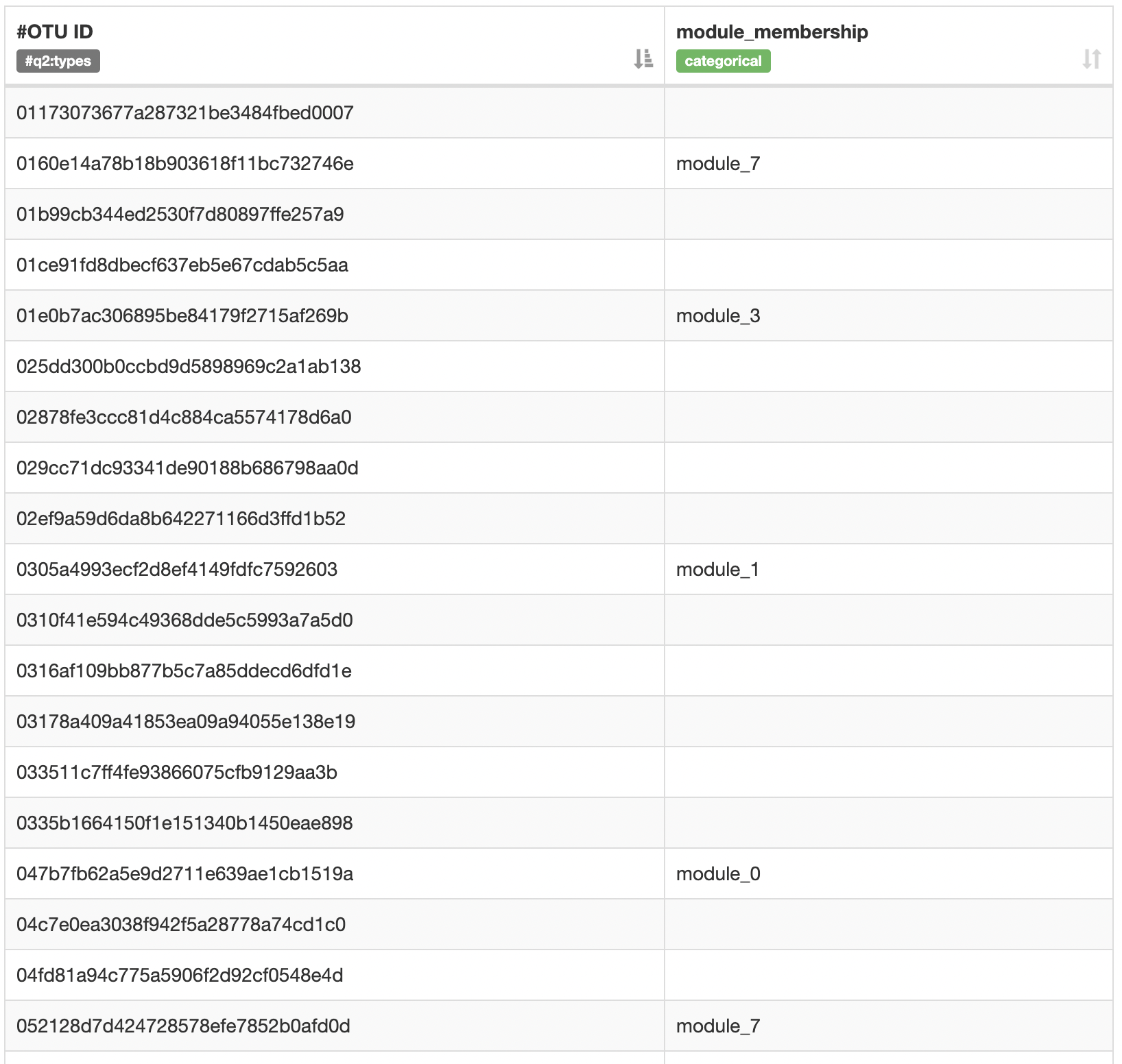
membership.qzaはqzvにすることでテーブルデータとして可視化ができる。
qiime metadata tabulate \
--m-input-file membership.qza \
--o-visualization membership.qzv
QIIME2 VIEWにてmembership.qzvを読み込むと以下のように表示される。
4. Cytoscapeでのネットワーク図の可視化
net.modules.qzaからネットワーク図として可視化できるgmlファイルを出力できる。
mkdir extracted-network
qiime tools extract --input-path net.modules.qza --output-path extracted-network/
出力されたnetwork.gmlをCytoscapeに読み込ませると可視化できる。
可視性のためのレイアウトには工夫が必要そう。
ASVだとQIIME2からランダム文字列を付加されるため、何がなんだかわからなくなるので、種をアサインした組成データでやるのが好ましい。
参考文献
SparCCの論文
Inferring Correlation Networks from Genomic Survey Data