腸内細菌と健康について
腸内細菌叢と疾患の関連は様々な研究から明らかになっている。生活習慣病から自己免疫疾患まで、宿主の生理機能に広範囲に影響を与えている。
ここ最近、メタゲノム解析など網羅的な解析技術の向上や大規模なコホート研究などによって、個別の疾患における腸内細菌叢の系統組成や多様性の特徴や変動性が明らかになってきており、腸内細菌叢の時系列観測を実施することで、疾患の前兆が腸内細菌叢により特定できる可能性が示唆されている。
宿主の健康状態を腸内細菌叢から調べるときにもっともわかりやすい方法は、健康に関連する微生物と疾患に関連する微生物のバランスを定量化することである。
この論文では、健康なグループと不健康なグループの間で頻度が高く検出される微生物種と、検出頻度が低い微生物種を比較した計算式を提案している。
Gut Microbiome Health Index (GMHI)について
筆者らは論文で腸内細菌叢の種レベルの系統組成情報に基づき、健康状態(診断された疾患の有無)を評価できるロバストな指標である"the Gut Microbiome Health Index (GMHI)"を開発している。
肥満や大腸腺腫、クローン病などの表現型を含む4,347もの糞便のショットガンメタゲノムシーケンスデータセットを用いて、健康状態の良い状態と悪い状態に関連する2種類の微生物種を特定しGMHIを作成しており、679サンプルのデータセットを用いて作成したモデルを検証している。
普及率をベースとした微生物種の特定戦略
まず、健康なヒト (healthy;H)と、不健康なヒト(nonhealthy;N)と定義したときに、それぞれH, Nにおける微生物種 (m)の普及率 (prevalence)を決定する。
p_{H,m}
p_{N,m}
次に、H, Nの普及率を比較するために、
- 普及率のfold change $f_m^{H,N}$
- 普及率の差分 $d_m^{H,N}$
の2つの基準を設けて、それぞれ以下のように定義する。
\frac{{p_{H,m}}}{{p_{N,m}}}
p_{H,m}-p_{N,m}
H, Nにおける有意な有病率の効果量は、両方の基準が普及率のfold change ($\theta_f$) と普及率の差分 ($\theta_d$) の最小の閾値を満たすような場合に存在すると考えられる。
f_m^{H,N} \ge \theta_f
d_m^{H,N} \ge \theta_d
を満たす、NよりもHで頻度が高く検出された微生物種"health-prevalent"を$M_H$ と定義している。
f_m^{N,H} \ge \theta_f
d_m^{N,H} \ge \theta_d
一方で上記を満たす検出可能なすべての種でNよりもHで頻度が低く検出された微生物種"health-scarce"を $M_N$ と定義している。
インストール方法
Github からスクリプトをダウンロードする。
Rスクリプトになっており、コンパイルなどは必要ない。
$ git clone https://github.com/jaeyunsung/GMHI_2020
$ cd GMHI_2020/
実行例
以下のRのバージョンを用いて実行した。
$ R --version
R version 4.0.4 (2021-02-15) -- "Lost Library Book"
各サンプルにおけるGMHIの算出
Github にて、公開されているテストデータとスクリプトを試す。
ちなみにGMHIを用いる際に、腸内細菌叢のショットガンメタゲノムシーケンスは、MetaPhlAn2で分類群のアサインメントをする必要がある。
最初にGMHI.Rを開いてパスを確認する。
以下のようにMH, MNの微生物群はすでに定義されており、species_relative_abundance_fileにMetaPhlAn2でアサインメントされたファイルを指定することで計算が可能だ。
分類群のファイルで検索するだけだったら、16S rRNAアンプリコンシーケンスでもいけるのかもしれない。種レベルまで分類されることは少ないけど。。。
species_relative_abundance_file <- "./species_relative_abundances.csv"
MH_species_file <- "./MH_species.txt"
MN_species_file <- "./MN_species.txt"
output_file = 'GMHI_output.csv'
tidyverseが必要なので、入っていない場合はRにてライブラリをインストールする。
install.packages("tidyverse")
# スクリプトの実行
$ Rscript GMHI.R
以下のように、各サンプルにおけるGMHIが算出された。
GMHI > 0 だと健康に分類され、GMHI <0 だと不健康に分類される。
| GMHI | |
|---|---|
| Sample1 | -0.631363762 |
| Sample2 | -2.345899453 |
| Sample3 | -2.705520798 |
| Sample4 | -1.217423513 |
| Sample5 | 1.74314722 |
| Sample6 | -0.294056833 |
| Sample7 | -3.740412863 |
| Sample8 | 0.454787851 |
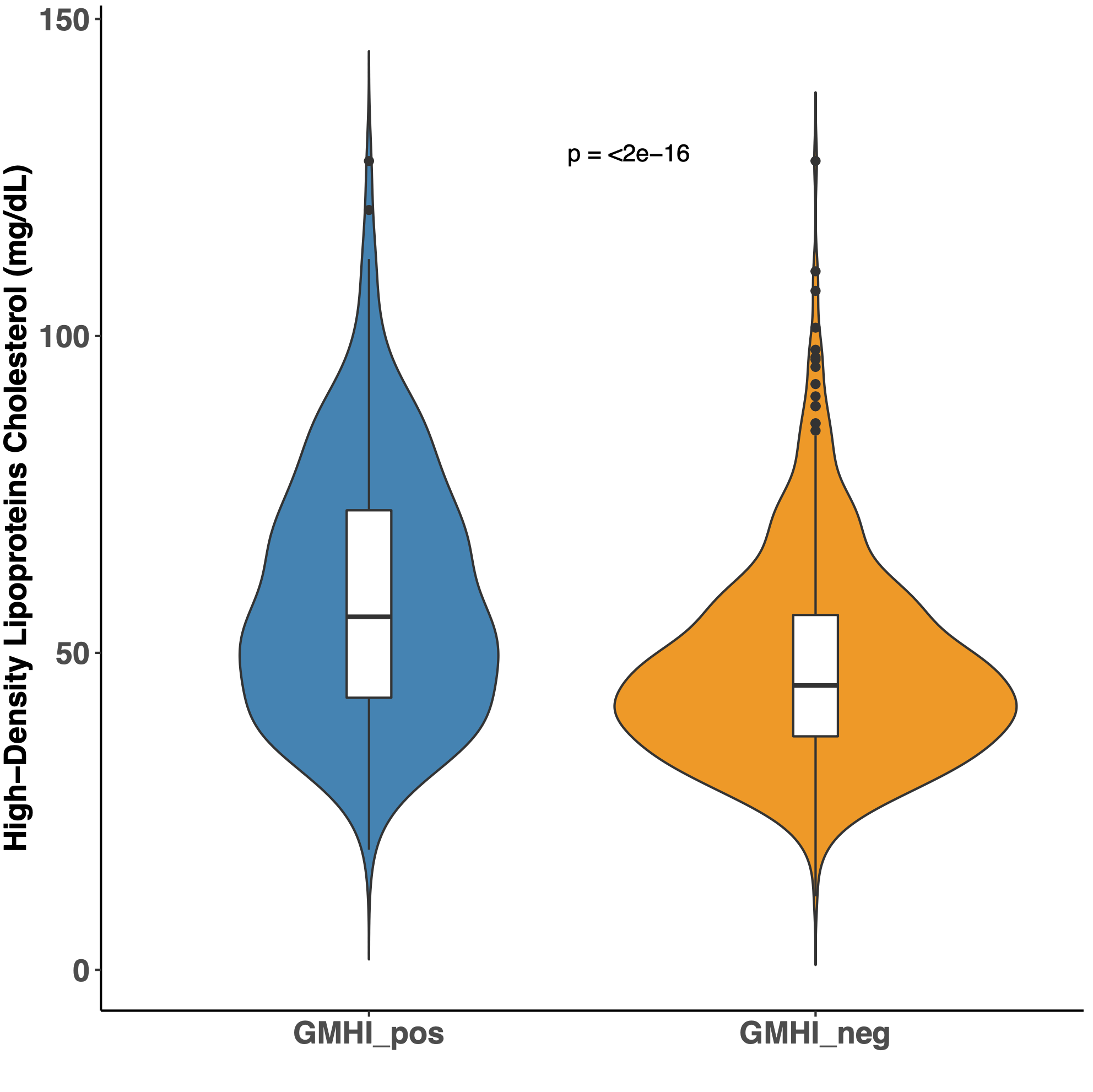
HDLコレステロールとGMHI
論文のFigure 2において、GMHIと善玉コレステロールと考えられるHDLコレステロールが正の相関を示していると報告されているが、これもgithubのスクリプトで計算と図表の描画ができる。
$ Rscript Fig2_TableS1_Table2.R
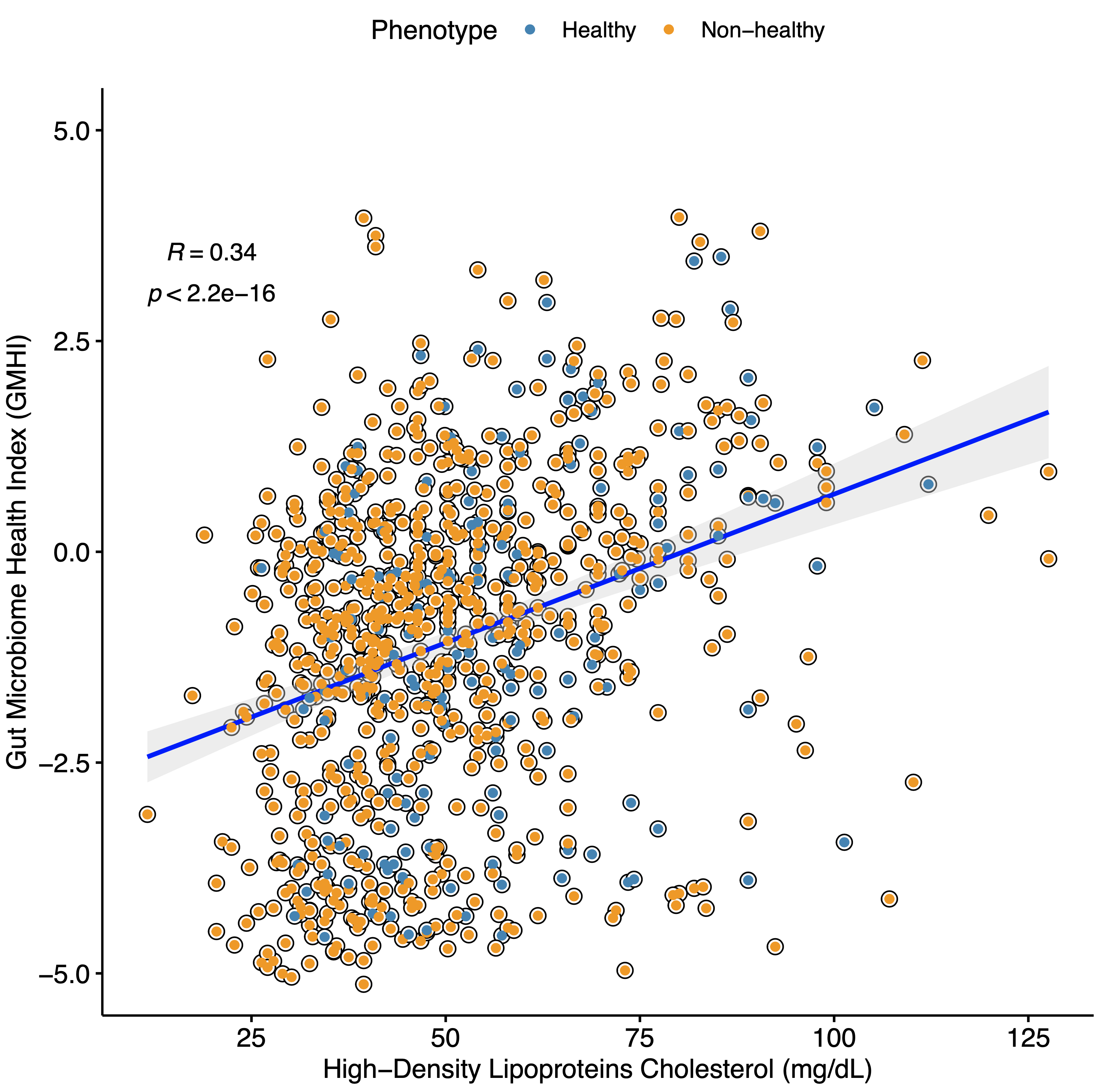
健康なグループ(GMHI> 0)は、不健康なグループ(GMHI < 0)において、HDLコレステロールの存在量が有意に多いことも示されている。