はじめに
情報化学系の論文にあるような**イケてるケミカルスペース (Chemical Space)**をPythonでどこまで再現できるか頑張ってみた話。
環境
- Windows 10
- RDKit 2020.09.3
- Mordred 1.2.0
- scikit-learn 0.22
- Jupyter Lab
表示したいこと
- 複数のデータセットを読み込み、主成分分析をした結果を表示したい
- データセット毎に色分けして表示したい
- ローディングプロットを表示したい
- 3Dで表示してみたい
準備
まず表示するデータセットを揃える。MoleculeNetおよびDrugBankから以下のデータセットの csv もしくは sdf ファイルをダウンロードしておく。
- FreeSolv
- ESOL
- Lipophilicity
- base
- BBBP
- DrugBank
プログラムで一括で読めるよう、以下のように各データセットの形式やSMILESのカラム名をtsv形式でまとめておく
dataset.csv
dataset name file_type file_name id_column smiles_column exp_column
FreeSolv csv SAMPL.csv iupac smiles expt
ESOL csv delaney-processed.csv Compound ID smiles measured log solubility in mols per litre
Lipophilicity csv Lipophilicity.csv CMPD_CHEMBLID smiles exp
base csv bace.csv CID mol pIC50
BBBP csv BBBP.csv name smiles p_np
DrugBank sdf structures.sdf DATABASE_ID SMILES EXACT_MASS
やってみよう
以下、Jupyter Labでケミカルスペースの表示をやっていく。
データの読み込み
import numpy as np
import pandas as pd
from rdkit import rdBase, Chem
from rdkit.Chem import AllChem, Descriptors
from rdkit.ML.Descriptors import MoleculeDescriptors
from mordred import Calculator, descriptors
# データセット定義ファイルの読み込み
df_dataset = pd.read_table("../datas/raw/datasets.csv", index_col=0)
# データセットの読み込み
mols = []
smiles_list = []
names_list = []
dataset_list = []
for row in df_dataset.iterrows():
if row[1]["file_type"] == "csv":
# CSVの読み込み
df_tmp = pd.read_csv("../datas/raw/" + str(row[1]["file_name"]))
smiles_tmp = df_tmp[row[1]["smiles_column"]].values
names_tmp = df_tmp[row[1]["id_column"]].values
for smiles, name in zip(smiles_tmp, names_tmp):
mol = Chem.MolFromSmiles(smiles)
if mol is not None:
mols.append(mol)
smiles_list.append(smiles)
names_list.append(name)
dataset_list.append(row[0])
else:
# SDFの読み込み
sdf_sup = Chem.SDMolSupplier("../datas/raw/" + str(row[1]["file_name"]))
for j, mol in enumerate(sdf_sup):
if mol is not None:
name = mol.GetProp(row[1]["id_column"])
if name is None or len(name) == 0:
name = (j+1)
smiles = Chem.MolToSmiles(mol)
mol = Chem.MolFromSmiles(smiles)
mols.append(mol)
names_list.append(name)
dataset_list.append(row[0])
RDKitの記述子計算
descriptor_names = [descriptor_name[0] for descriptor_name in Descriptors._descList]
descriptor_calculator = MoleculeDescriptors.MolecularDescriptorCalculator(descriptor_names)
rdkit_descriptors_results = [descriptor_calculator.CalcDescriptors(mol) for mol in mols]
df_rdkit = pd.DataFrame(rdkit_descriptors_results, columns = descriptor_names, index=names_list)
# 数値変換後のnullの列チェック
df_rdkit = df_rdkit[df_rdkit.columns[~df_rdkit.isnull().any()]]
Modredの記述子計算
# mordredの記述子計算
mordred_calculator = Calculator(descriptors, ignore_3D=True)
df_mordred = mordred_calculator.pandas(pd.Series(mols, index=names_list))
# 数値型への変換
for column in df_mordred.columns:
if df_mordred[column].dtypes == object:
#print("{0}/{1}".format(column, df_mordred[column].dtypes))
df_mordred[column] = df_mordred[column].values.astype(np.float32)
else:
#print("{0}/{1}".format(column, df_mordred[column].dtypes))
pass
# 数値変換後のnullの列チェック
df_mordred = df_mordred[df_mordred.columns[~df_mordred.isnull().any()]]
データのまとめ
# RDKit, Mordredでヘッダをつける
for df_tmp, prefix in zip([df_rdkit, df_mordred], ["RDkit.", "Mordred."]):
columns_new = [(prefix + column_tmp) for column_tmp in df_tmp.columns]
df_tmp.columns = columns_new
df_all = pd.concat([df_rdkit, df_mordred], axis=1)
記述子の削減
このままだと記述子が多いので削減する。
# 分散が0のものを除去
from sklearn.feature_selection import VarianceThreshold
tmp_values = df_all.values
select = VarianceThreshold()
tmp_values = select.fit_transform(tmp_values)
df_all_vt = df_all[df_all.columns[select.get_support()]]
# 相関の高いものを除去
def corr_column(df, threshold):
df_corr = df.corr()
df_corr = abs(df_corr)
columns = df_corr.columns
# 対角線の値を0にする
for i in range(0, len(columns)):
df_corr.iloc[i, i] = 0
while True:
columns = df_corr.columns
max_corr = 0.0
query_column = None
target_column = None
df_max_column_value = df_corr.max()
max_corr = df_max_column_value.max()
query_column = df_max_column_value.idxmax()
target_column = df_corr[query_column].idxmax()
if max_corr < threshold:
# しきい値を超えるものがなかったため終了
break
else:
# しきい値を超えるものがあった場合
delete_column = None
saved_column = None
# その他との相関の絶対値が大きい方を除去
if sum(df_corr[query_column]) <= sum(df_corr[target_column]):
delete_column = target_column
saved_column = query_column
else:
delete_column = query_column
saved_column = target_column
# 除去すべき特徴を相関行列から消す(行、列)
df_corr.drop([delete_column], axis=0, inplace=True)
df_corr.drop([delete_column], axis=1, inplace=True)
return df_corr.columns
non_corr_columns = corr_column(df_all_vt, 0.90)
df_all_ct = df_all_vt.loc[:, non_corr_columns
オートスケーリング
# オートスケーリング
from sklearn import preprocessing
from sklearn.preprocessing import StandardScaler
ss = StandardScaler()
train_X_scaled = ss.fit_transform(df_all_ct.values)
主成分分析
# 主成分分析を行う
from sklearn.decomposition import PCA
pca_model = PCA(n_components=3)
Z = pca_model.fit_transform(train_X_scaled)
いよいよ可視化
2Dでの可視化をしてみよう。
ローディングプロットは、第一成分、第二成分それぞれで絶対値の大きなもの5つを表示した。
%matplotlib inline
import matplotlib.pyplot as plt
plt.style.use('seaborn')
figure = plt.figure(figsize=(12,10))
ax_pca = figure.add_subplot(111)
dataset_list = np.array(dataset_list)
for dataset, color in zip(np.unique(dataset_list), plt.rcParams['axes.prop_cycle'].by_key()['color']):
Z_dataset = Z[np.array(dataset_list)==dataset]
ax_pca.scatter(Z_dataset[:, 0], Z_dataset[:, 1], c=color, alpha=0.3, label=dataset)
ax_pca.set_title("PCA Result")
ax_pca.set_xlabel("PCA 1")
ax_pca.set_ylabel("PCA 2")
plt.legend(bbox_to_anchor=(1, 1), loc='upper right', borderaxespad=1, fontsize=12)
indexes_0 = np.abs(pca_model.components_[0]).argsort()[-5:][::-1]
indexes_1 = np.abs(pca_model.components_[1]).argsort()[-5:][::-1]
# ローディングプロット
keisu = 100
for index in indexes_0:
name = df_all_ct.columns[index]
value_0 = pca_model.components_[0][index] * keisu
value_1 = pca_model.components_[1][index] * keisu
plt.text(value_0, value_1, name)
ax_pca.arrow(x=0,y=0,dx=value_0,dy=value_1,width=0.01,head_width=0.5,head_length=0.5,length_includes_head=True,color='k')
for index in indexes_1:
name = df_all_ct.columns[index]
value_0 = pca_model.components_[0][index] * keisu
value_1 = pca_model.components_[1][index] * keisu
plt.text(value_0, value_1, name)
ax_pca.arrow(x=0,y=0,dx=value_0,dy=value_1,width=0.01,head_width=0.5,head_length=0.5,length_includes_head=True,color='k')
こんな感じになった。
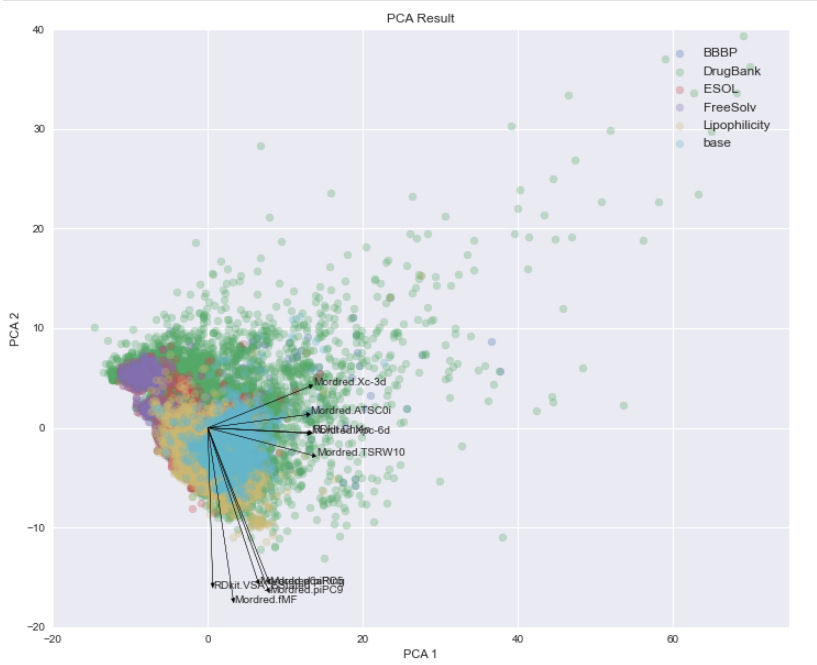
続いて3Dで表示してみよう。3Dの場合、テキストの表示がうまくいかなかったためローディングプロットは外している。
# 主成分分析結果を可視化する
%matplotlib inline
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
plt.style.use('seaborn')
figure = plt.figure(figsize=(15,12))
ax_pca = figure.add_subplot(111, projection='3d')
dataset_list = np.array(dataset_list)
for dataset, color in zip(np.unique(dataset_list), plt.rcParams['axes.prop_cycle'].by_key()['color']):
Z_dataset = Z[np.array(dataset_list)==dataset]
ax_pca.scatter(Z_dataset[:, 0], Z_dataset[:, 1], Z_dataset[:, 2], c=color, alpha=0.3, label=dataset)
ax_pca.set_title("PCA Result")
ax_pca.set_xlabel("PCA 1")
ax_pca.set_ylabel("PCA 2")
ax_pca.set_zlabel("PCA 3")
ax_pca.set_xlim([-10, 20])
ax_pca.set_ylim([-10, 20])
ax_pca.set_zlim([-10, 20])
plt.legend(bbox_to_anchor=(1, 1), loc='upper right', borderaxespad=1, fontsize=12)
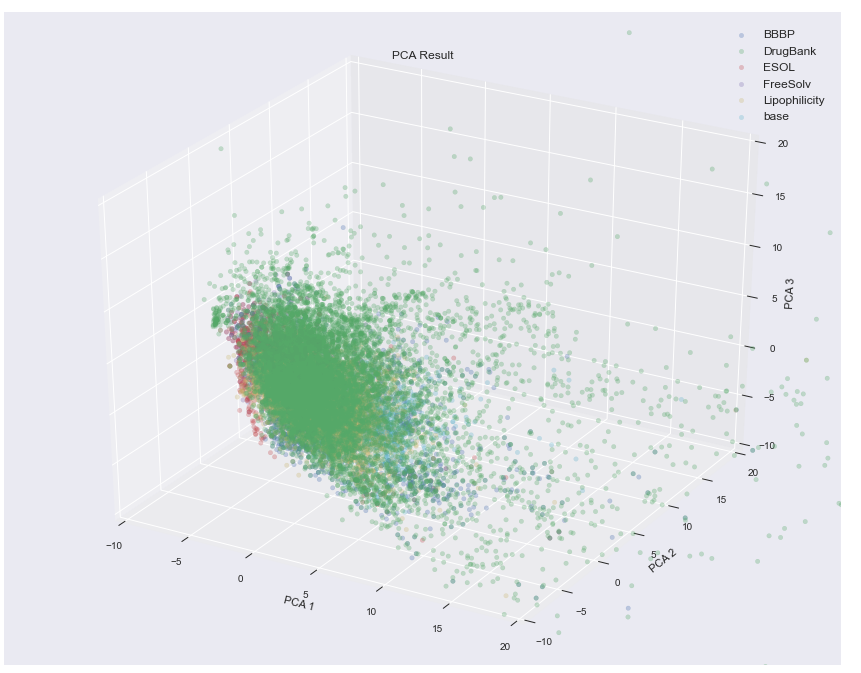
見やすいかというと微妙かもしれない。
表示結果に対する感想
- DrugBankの化合物の空間に対し、MoleculeNetの各データセットの空間が意外と狭い
- データセット毎に化合物空間が固まっている傾向がみられる。その中でDrugBankは件数が多いこともあるが幅広く散らばっている。
- ローディングプロットとして表示した記述子の意味が分かりにくい。LogP、LogD、pKa、分子量、The number of rotatable bonds、he number of aromatic atoms/bonds, Topological polar surface area, hydrogen bond acceptor/donor 等、分かりやすい記述子のみ使って主成分分析をした方が解析しやすいかもしれない。
表示の課題
課題は以下の通り。
- ローディングプロットの線や文字が重なってみずらい
- 拡大/縮小をしたい。
- 3Dグラフについては見る角度を変えてみたい。