はじめに
分子が標的タンパク質にどのように結合するかを予測することは、創薬の重要な課題である。これまで様々なドッキングシミュレーション手法が提案されてきたが、最近この分野にも機械学習の波が押し寄せて来ているのではないかと思い調べた結果、EquiBindという手法を見つけ、実際に予測を試してみたので共有したい。
環境
こんな感じ。他にもファイル操作のためドッキングシミュレーションソフトがあればよい。
- Windows10
- PyMol
- RDKit
- Jupyter Lab
本記事で試したこと
手順はHira Laboさんの EquiBind (深層学習によるタンパク質-リガンドドッキング予測)のインストール•使い方 をそのまま参考とさせていただいた。ただ、Hira Laboさんの手順では、入力に与えるリガンドの座標として、タンパク質と結合しているPDBの座標をそのまま使っており、本当に予測を行えているか疑問が残ったので、本記事では 予測前にタンパク質とリガンドが離れていることを確認する という手順を盛り込んだ。
やってみよう
予測データの準備
今回はSARS-Cov-2 3C-like proteinaseと既存化合物との相互作用について調べてみる。
PDBファイルの準備
まず、Protein Data Bank (PDB ID:6LU7) から PDBファイルをダウンロードしよう。
PDBファイルからタンパク質とリガンドを抽出
このファイルには、タンパク質(SARS-Cov-2 3C-like proteinase)とリガンド(ペプチド模倣阻害剤)が結合した状態で含まれているので、ドッキングソフトウェア等で、タンパク質をPDBファイルに、リガンドをsdfファイルに保存しよう。
リガンドに水素を付加し、3次元構造を立ち上げ
Jupyter Labを起動し、以下の手順でリガンド化合物の水素付加、3次元構造立ち上げを行う。
まず、モジュール読み込もう。
from rdkit import Chem
from rdkit.Chem import AllChem
ligand_mol = Chem.MolFromMolFile("./corona/6lu7_ligand.sdf")
SDFファイル読み込もう。
ligand_mol = Chem.MolFromMolFile("./corona/6lu7_ligand.sdf")
水素を付加しよう。
ligand_mol = Chem.AddHs(ligand_mol, addCoords=True)
3次元構造を立ち上げよう。
AllChem.EmbedMolecule(ligand_mol, AllChem.ETDG())
座標を生成しなおしたことによって、PDBファイルの座標が上書きされるはずだ。
ファイルに上書き保存しよう
writer = Chem.SDWriter("./corona/6lu7_ligand.sdf")
writer.write(ligand_mol)
writer.close()
他にも本タンパク質の結合親和性が高いとされるロピナビルやリトナビルについてもmolファイルを入手し、同様に用意しておこう。EquiBindがmol拡張子に対応しているかは分からないが、sdfにしておくと無難だ。
正解結合位置の確認
元のPDBファイルをPyMolで表示し、おおよその結合位置を確認してみよう。
pymol 6lu7.pdb
ちょっと分かりにくいが大体下図の位置になる。
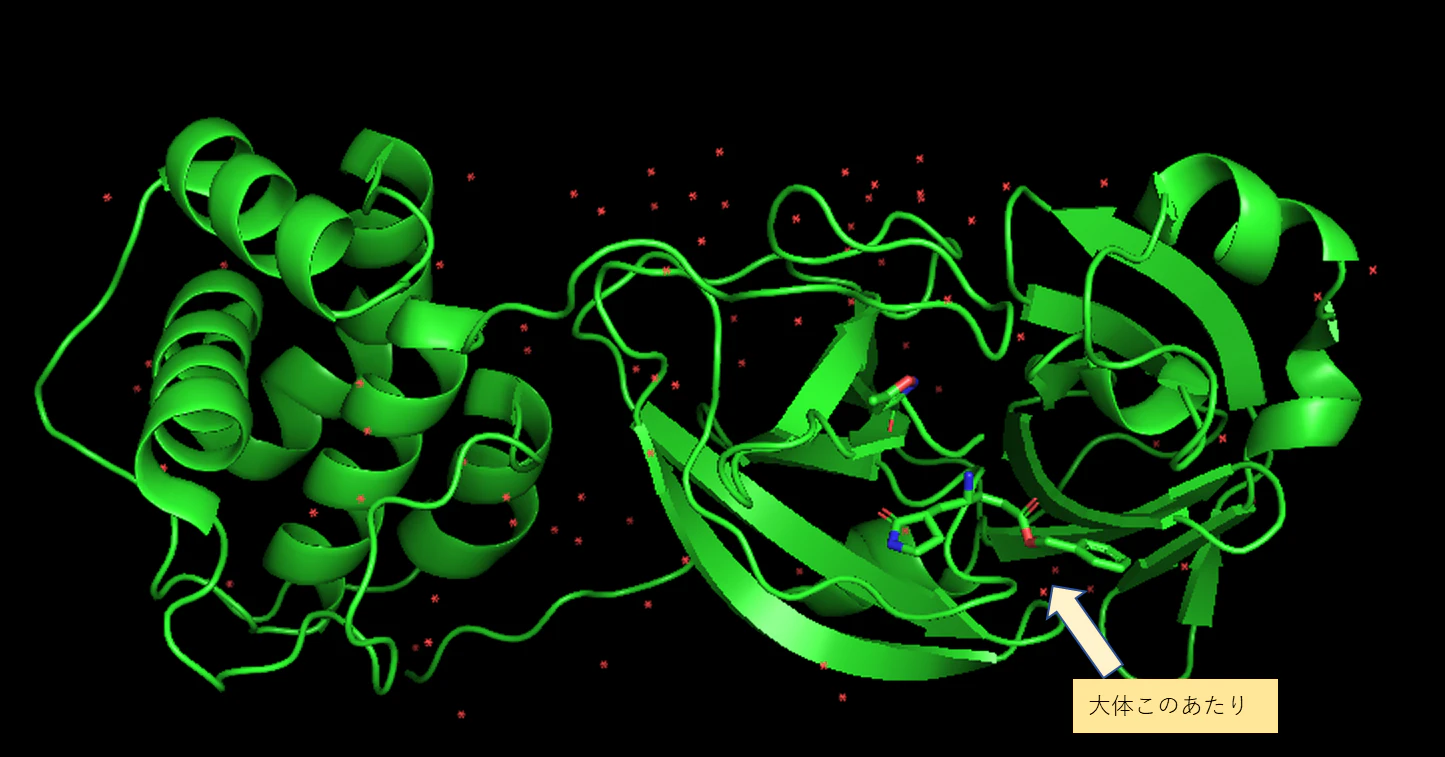
予測前のタンパク質とリガンドの位置関係の確認
続いてPyMolで、タンパク質とリガンドの配置を確認してみよう。
EquiBindのインストールフォルダの下の .\data\inputs\corona1\ の下にタンパク質ファイル、リガンドファイルをそれぞれ6lu7_protein.pdb、6lu7_ligand.sdfというファイル名で置いておく。 2番目, 3番目の化合物も、corona1のところをcorona2, corona3に変更して同様に保存しよう。
次に以下の通り、タンパク質とリガンドのファイルをPyMolで確認してみよう。
pymol data\inputs\corona1\6lu7_protein.pdb data/inputs/corona1/6lu7_ligand.sdf
下のようにタンパク質とリガンドが離れていることが確認できる。
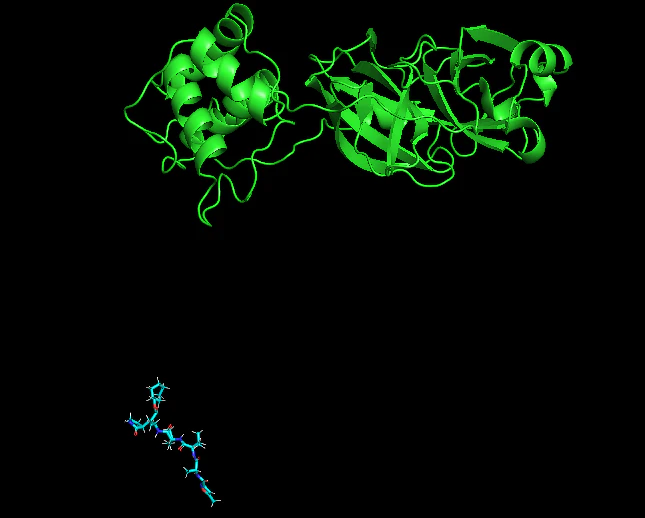
残り2つの化合物についても離れているか確認してみよう。これで予測の準備は整った。
予測実行
さてEquiBindのインストールフォルダに移動し、以下のコマンドで予測してみよう。ymlの設定等は、Hira Laboさんのページを参照してほしい。
python inference.py --config=configs_clean/inference.yml
これで3つの化合物の予測後の座標が格納されたsdfファイルが以下に出力されるはずだ。
data\results\output\corona1\lig_equibind_corrected.sdfdata\results\output\corona2\lig_equibind_corrected.sdfdata\results\output\corona3\lig_equibind_corrected.sdf
予測は数秒で終わる。従来手法に比べた本手法の大きなメリットの1つだろう。
予測後の確認
3つの化合物を順次見ていこう。
ペプチド模倣阻害剤
このコマンドでタンパク質とリガンドのドッキングの予測結果を可視化できる。
pymol data\inputs\corona1\6lu7_protein.pdb data/results/output/corona1/lig_equibind_corrected.sdf
こんな感じ。どこまでの精度だとOKなのかが良く分からないが、正解例と割と近いのではないだろうか。
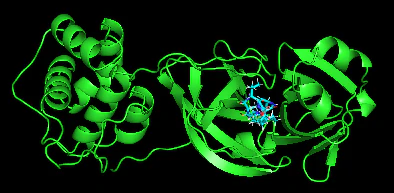
ロピナビル
続いて2番目の化合物。1番目と同様の結果だ。
pymol data\inputs\corona2\6lu7_protein.pdb data/results/output/corona2/lig_equibind_corrected.sdf
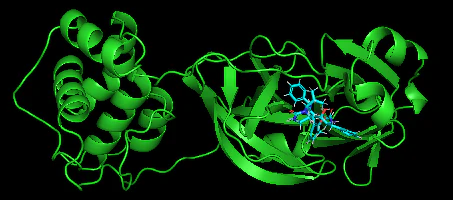
リトナビル
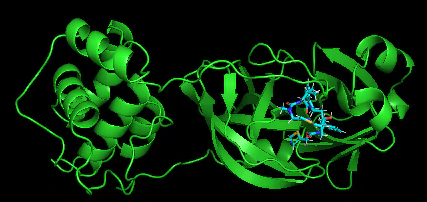
続いて3番目の化合物。これも1番目と同様の結果だ。
pymol data\inputs\corona3\6lu7_protein.pdb data/results/output/corona3/lig_equibind_corrected.sdf
感想
タンパク質データの取り扱いに不慣れなため、もっと分かりやすい可視化方法があるかもしれない。ともかく、ドッキングシミュレーションのど素人の自分でも予測まではできた。本手法はこの分野への深層学習適用の第一歩にすぎず、これからどんどん改良されたモデルが出てくるような気がする。これからも目が離せない!