サマリー
オープンソースのPoly Peak Parserを使い、サンガー法の出力ファイル(.ab1または.scf)とリファレンス配列から、ヘテロ接合Indel(挿入・欠失)やヘテロ接合SNVを確認します。CRISPRやTALENでも活躍します。
オンラインにファイルを流せる場合はこちらで利用可能です。使用感が確認できます。
Mutation Surveyorとは
サンガー法の結果解析として用いられている変異解析ソフトウェアです。独自のアルゴリズムにより、高精度でヘテロ接合Indelやヘテロ接合SNVを検出できます。ただし、破茶滅茶にお高い(数十〜数百万円)です。
Poly Peak Parserとは
その名の通り、クロマトグラムに複数のピークが重複して存在するサンガー法の結果を解析できるRパッケージです。2014年にユタ大学のJonathon T. Hillらによって発表されました*。Mutation Surveyorが手元にない...ヘテロ接合のIndelを確認するのが面倒...そんなときに役立ちます。
*Hill, Jonathon T., et al. "Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products." Developmental Dynamics 243.12 (2014): 1632-1636.
環境
Version 4.0 以降のR環境があれば使えます。プラットフォームはWindowsでも、Macでも、Linuxでも大丈夫です。
$ R --version
R version 4.0.3 (2020-10-10) -- "Bunny-Wunnies Freak Out"
Copyright (C) 2020 The R Foundation for Statistical Computing
Platform: x86_64-apple-darwin17.0 (64-bit)
導入
びっくりするほど簡単。公式のインストール方法に沿っていくだけです。Rのコンソールを開いて以下のコマンドを打っていきます。確認画面が出てきたら問題がないことを確認し、AllかYesを選択します。
> if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
> BiocManager::install("sangerseqR")
> browseVignettes("sangerseqR")
これで完了です。
使ってみる
まずはライブラリーを読み込み、PolyPeakParserを起動します。
> library(sangerseqR)
> PolyPeakParser()
ゴニョゴニョといろいろなものが読み込まれていきます。しばらくすると自動でブラウザが立ち上がり、以下のような画面が出てきます。
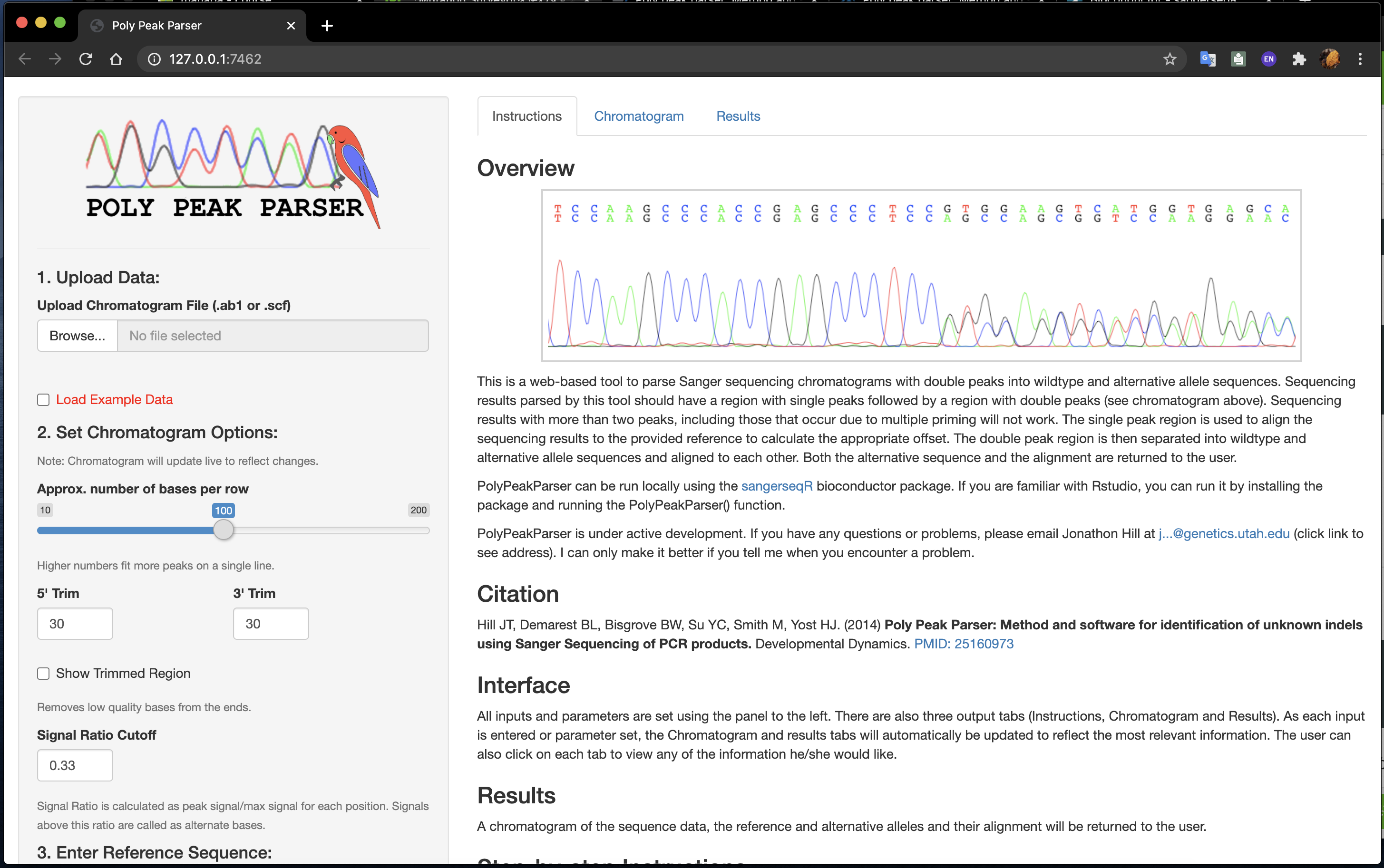
左上のボタンBrowseから解析対象のファイルを選択すると、自動的に処理が始まります。処理が終わると自動的にクロマトグラム表示になります。今回はこちらで公開されている無料のab1ファイルA_forward.ab1を例に解説します。
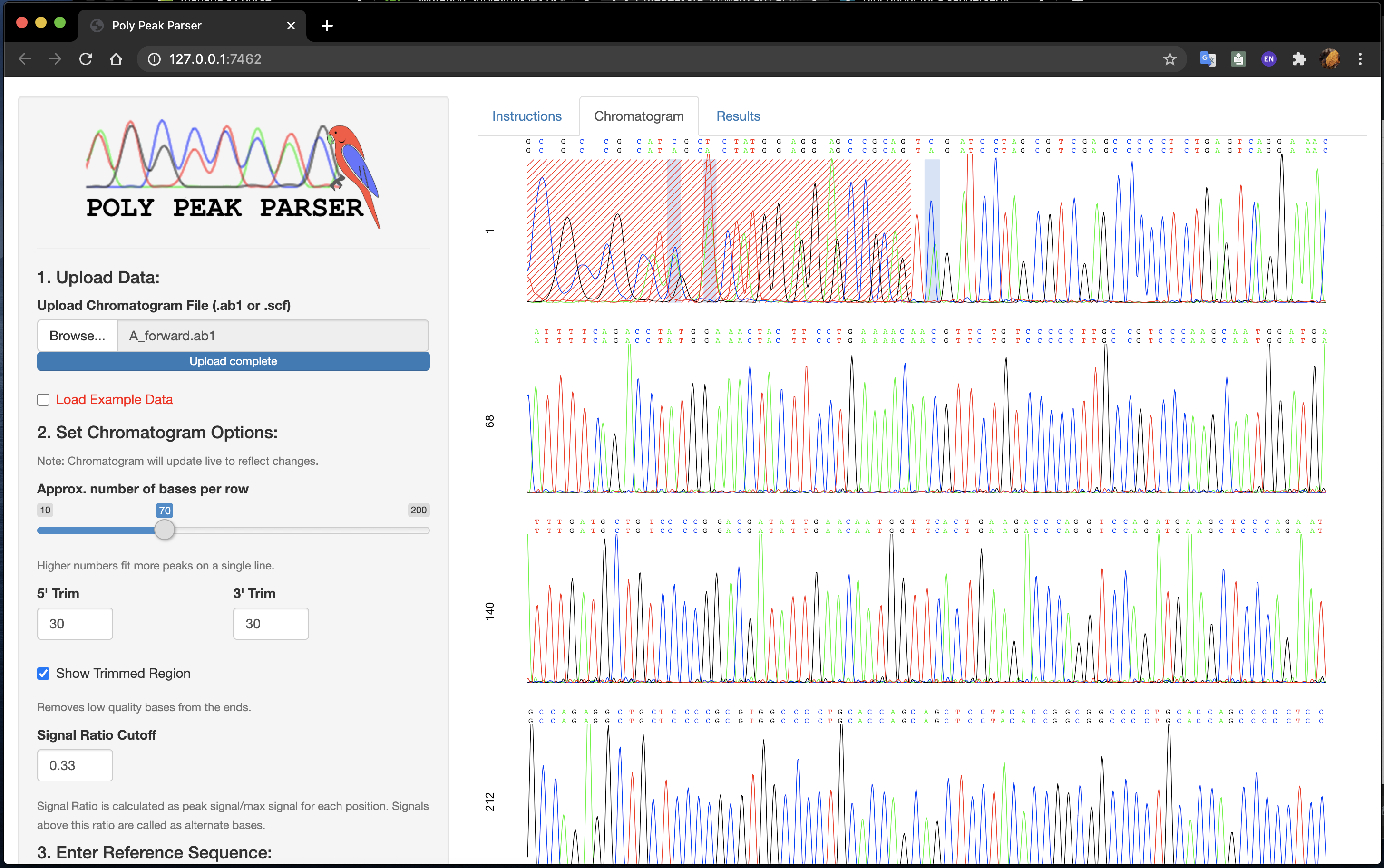
画面左部のペインでかんたんな表示設定や解析設定ができます。
Approx. number of bases per row : クロマトグラム1行あたりの塩基数
5' Trim : 5末端のトリミング長
3' Trim : 3末端のトリミング長
Show Trimmed Region : トリミングされた配列を赤斜線で潰すかどうか
Signal Ratio Cutoff : いちばん大きい波形に対してどれほどの比率の大きさの2nd Peakを拾ってくるか
また、Enter Reference SequenceにEnsemblなどで拾ってきたリファレンス配列を入れてあげれば、2nd Peak(2番めに高い波形)との比較ができます。2nd Peakがない場合、1st Peakが採用されます。
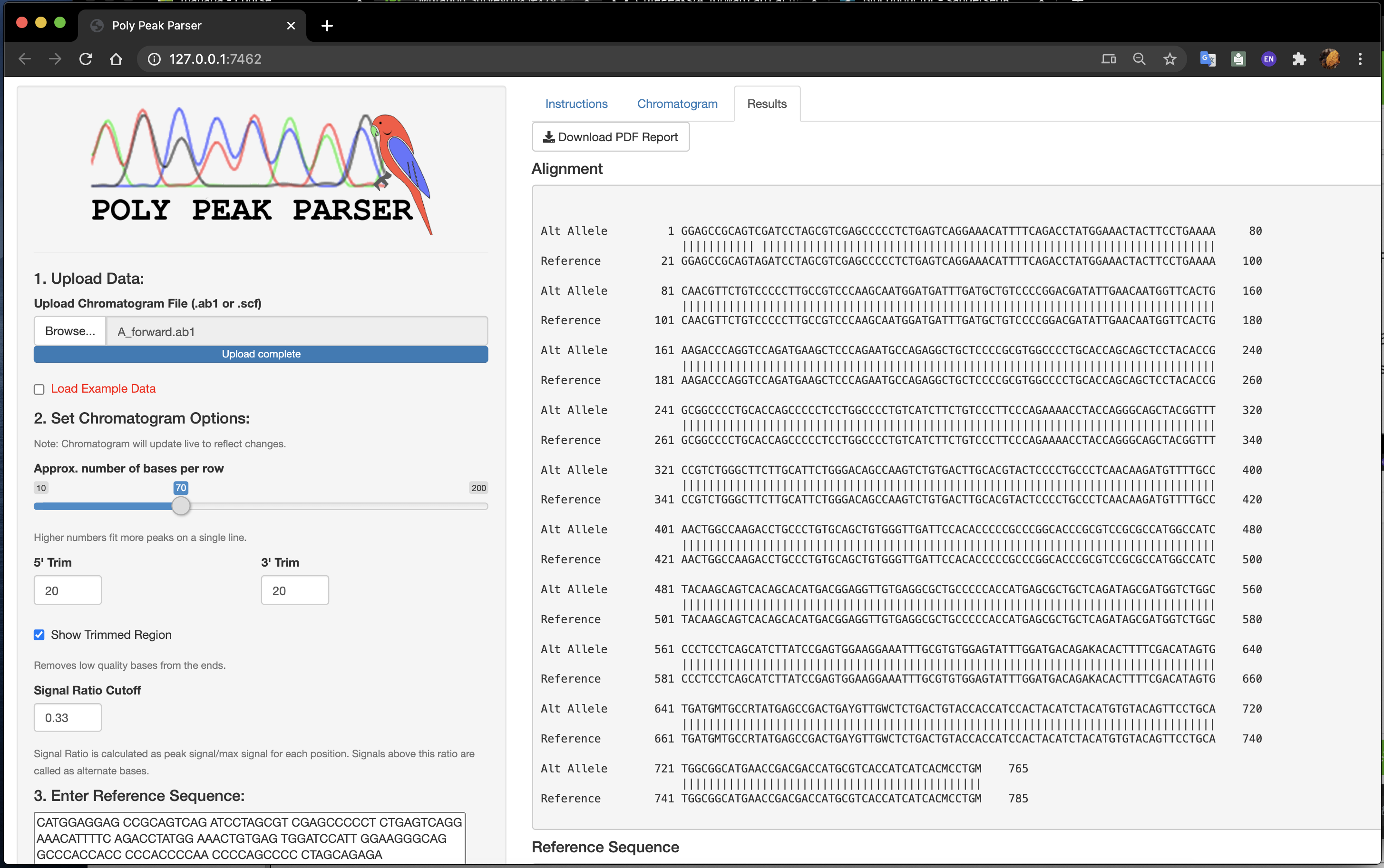
リファレンス配列と合致する場所には|が表示されます。今回の例では1箇所だけヘテロ接合と思われる変異が発見されました。
 |
|---|
こんな感じで、もう少し長く連続したヘテロ接合のIndelがあるサンプルのab1ファイルを見てみると、これまでは手作業で行っていたであろう「野生型でない方の配列をリファレンス配列と比べてIndel長を検討する」という作業が数秒で行えるようになります。(そういったファイルは持っているのですが、大人の事情でここではお示しできません...すみません)
まとめ
有料ソフトほどではないですが、ヘテロ接合mutationの検出にかかる時間を短縮することに成功しました。他にも機能があるライブラリーなので、詳しくはBioconductorのページからReference Manualをご覧ください。