概要
本稿では,機械学習の技術を用いて原子間ポテンシャルを作成する方法を紹介します.このような試みはいろいろありますが,本稿では「ニューラルネットワーク」を用いて原子間ポテンシャルを作成します (参考文献:J. Behler and M. Parrinello, Phys. Rev. Lett. 98, 146401 (2007)) ニューラルネットワークポテンシャルを作成するためには教師データが必要ですが,これは第一原理分子動力学シミュレーションを用いて作成します.さらに,作成したニューラルネットワークポテンシャルを用いて分子動力学シミュレーションを実行する方法も説明します.ここで説明する作業の入出力データはこのGitHubのレポジトリーにあるので,興味があればぜひ試してみてください.
アプリケーションのインストール
本稿の作業ではいろいろなアプリケーションを用いて目的の作業を行う予定です.先のGitHubのレポジトリーのデータを用いるのであれば不要なものもありますが,ご興味に応じてインストールして作業を行ってみてください.
- atomsk 原子配置の編集や変換を行うことができるツールです.
- PHASE/0 第一原理計算を行うことができるソフトウェアです.教師データ作成に使います.
- ænet 機械学習ポテンシャルを作成することのできるソフトウェアです.機械学習ポテンシャル作成に使います.
- LAMMPS 古典分子動力学シミュレーションを行うことができるソフトウェアです.検証の分子動力学シミュレーションに使います.
- Jmol 原子配置を可視化することのできるソフトウェアです.可視化に使います.
PHASE/0のインストール
このサイトの情報などを参考にしてインストールしてください.
ænetのインストール
ænet本体のビルド
まずはænetのダウンロードページからtar ballをダウンロードします.このページからダウンロードできる最新版は aenet-2.0.3.tar.bz2 のようです.適当な位置にコピーし,展開します.
cd $HOME
tar -jxvf aenet-2.0.3.tar.bz2
まずはLBFGSライブラリーをビルドします.これはlibディレクトリーにおいて行います.
cd aenet-2.0.3
cd lib
make F90='ifort -c'
F90のあとには利用したいFotran90コンパイラー(とオブジェクトファイル生成のみを行うことを伝えるオプション)を指定します.デフォルト値はgfortran -cです.そのあとはsrcディレクトリーにおいてmakeを実行します.
cd ../src
make -f makefiles/Makefile.ifort_intelmpi
srcディレクトリーの下のmakefilesディレクトリーにコンパイラーやMPI別のmakefileが配置されています.お使いのシステムにインストールされているコンパイラーおよびMPIに応じて適切なmakefileを選んで実行してください.また,BLASおよびLAPACKが必要なようです.MKLやOpenBLAS用のmakefileが用意されているようです.上述の例ではIntel FortranコンパイラーおよびIntel MPIを用いるmakefileを選んでmakeコマンドを実行しました.この組み合わせの場合MKLがインストールされていおり,それを利用することが前提となっているようです.お使いのシステムにおいてどうしても対応するmakefileが存在しない場合は似たものを選び,編集する必要があります.うまくいけばbinディレクトリーにgenerate.x-2.0.3-xxx, predict.x-2.0.3-xxx, train.x-2.0.3.xxx などの実行可能ファイルが作成されます.ここでxxxはコンパイラーやMPIの組み合わせを表す文字列です(たとえば ifort_intelmpi )以降,単にgenerate.x train.x predict.x などと記述することにします.
ænetのLAMMPSインターフェースのインストール
ænetには単体でLAMMPSと組み合わせて利用する方法は用意されていません.そこで Hideki MORIさんによるLAMMPSインターフェースを用います.このインターフェース配布元の指示に従ってインストールします.なお,インターフェースは用いたいLAMMPSのバージョンと対応する版をreleasesから探し,ダウンロードする必要があります.ここではLAMMPSの27May2021版を用いたので,インターフェースも2021May27版を取得しました.
cp -r aenet-lammps/USER-AENET/ lammps-27May2021/src/
cp -r aenet-lammps/aenet lammps-27May2021/lib
cp aenet-2.0.3.tar.bz2 lammps-27May2021/lib/aenet/
cd lammps-27May2021/lib/aenet
tar -jvxf aenet-2.0.3.tar.bz2
patch -u -p1 -d aenet-2.0.3/ < aenet-2.0.3_lammps.patch
cd aenet-2.0.3/src/
make -f makefiles/Makefile.gfortran_serial lib
cd ../../../../src
make yes-user-aenet
make mpi
以上の手続きがエラーなく終了すればænetを用いることのできるLAMMPSバイナリーlmp_mpiがLAMMPSのsrcディレクトリーの下に作成されます.
教師データの作成
機械学習ポテンシャルを作成するためには,まずは教師データを作成する必要があります.教師データとなるのは原子座標と対応するエネルギーおよび原子間力です.これらのデータは通常第一原理計算を用いて作成します.本稿では,第一原理バンド計算プログラムPHASE/0を用いて教師データを作成しようと思います.対象は$\alpha$-quartz (${\rm SiO}_2$) にします.
計算モデル作成
まずは入力となる結晶構造を作成します.$\alpha$-quartzの結晶構造は六方晶で,単位胞に9個の原子が含まれます.このサイズでは多様な原子配置をサンプルすることができないので,少し拡大します.具体的には以下の方針で拡大します.
- 六方晶を直方晶に取り直す
- 結果得られる結晶の$a$軸と$b$軸をそれぞれ2倍にする
この操作によって原子数はもとの8倍になり,72原子の結晶が得られます.
CIFの入手
CIFとはCrystallographic Information Fileの略で,結晶構造を記述するデファクトスタンダードのフォーマットです.$\alpha$-quartzのCIFを入手します.ありふれた結晶なので入手経路はいろいろあると思いますが,たとえばAmerican Mineralogist Crystal Structure DataBaseから手に入れることができます.もしくは単純にquartz CIFというキーワードから到達することもできるかもしれません.
CIFに原子を加える
CIFは余計な情報を含んでいません.そのため,対称性で等価な原子は記録されていません.これから用いるツール群はこれを理解できるものばかりではないため,まずは「対称性から等価な原子も記述されている座標データファイル」を作成します.この操作はatomskの以下のコマンドによって実現することができます.
atomsk quartz.cif quartz.xsf
出力先としてはxsf形式を採用しました.これは,atomskの出力するCIFは形式が特殊で通常のプログラムは扱えないことが多いためです.
結晶を拡大する
上で述べたように,入手したCIFの構造をまずは六方晶から直方晶に取り直します.この作業は
- $a$軸を2倍にしたスーパーセルを作成する.結果はカルテシアン座標で保存する
- 結果得られる結晶の$b$軸の$x$座標を0にする
- セルからはみ出した原子が気持ち悪く感じる場合,それを単位胞内に押し込める
という流れで実現することができます.合間合間で発生する座標変換はPHASE/0に付属するconv.pyというスクリプトを用いることにします.conv.pyは対話的に用いることができる座標データコンバーターです.その使い方の詳細はPHASE/0のマニュアルをご覧ください.
1.の作業はconv.pyに--na=2というオプションをつけて起動することによって実現することができます.出力形式はカルテシアン座標系であるxsfを選んでください.
$HOME/phase0_2022.01/bin/conv.py --na=2
...
ここでPHASE/0の2022.01版がホームディレクトリーにインストールされていると仮定しました.結果得られるcoord.xsfの内容は以下のようになっているはずです(CIFの入手元によって若干違うかもしれません)
CRYSTAL
PRIMVEC
9.832 0.0 0.0
-2.458 4.25738089 0.0
0.0 0.0 5.4054
PRIMCOORD
18 1
Si 2.3090452 0 0
O 1.3767258 1.13629496 0.64378314
Si -1.1545226 1.9996918 3.6054018
O 0.2956974 1.760427 2.96161866
O 3.2435768 0.62413204 4.24918494
Si 1.3034774 2.25768908 1.8054036
...
...
3行目から5行目が単位胞の指定です.4行目の$b$軸を表すベクトルの$x$座標を,エディターなどを用いて-2.458から0.0に変更しましょう.その結果直方晶の結晶を得ることができます.この結晶に対し,さらに$b$軸と$c$軸をそれぞれ2倍にします.ついでに単位胞の外に存在する原子を単位胞内に押し込めることを行います.これもconv.pyを用いて行います.以下の要領でconv.pyを起動してください.変換先のファイル形式はとりあえずなんでもよいですが,汎用性の高いCIFなどが推奨です.
$HOME/phase0_2022.01/bin/conv.py --nb=2 --nc=2 --pack
この操作の結果得られるCIFファイルをJmolで可視化した結果が次の図です.
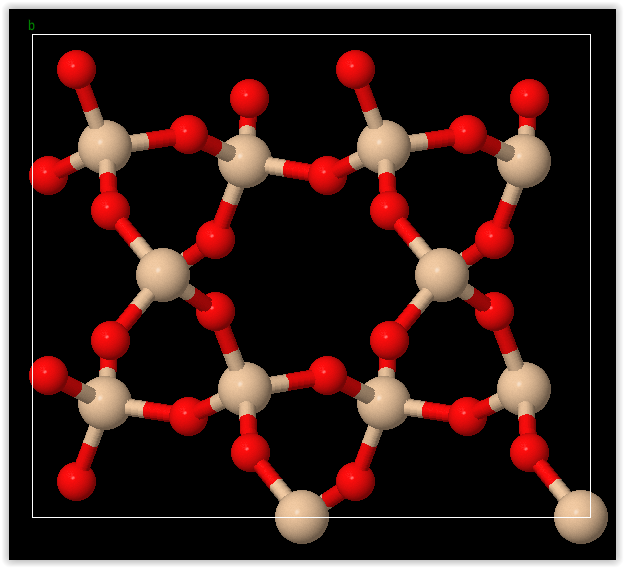
第一原理分子動力学シミュレーション
PHASE/0を用いた第一原理分子動力学シミュレーションを実行します.PHASE/0の使い方についての説明はあまりに長くなるので,以降の説明はある程度習熟しているものとしてすすめます.詳しくない場合はPHASE/0のマニュアルやチュートリアルなどを参照するようにしてください.
入力パラメーターファイル
前節の作業によって座標データファイルが得られました.conv.pyの出力先としてPHASE/0の入力を選ぶことによってPHASE/0の入力形式の座標データを作成します.得られるファイルnfinp.dataは以下のような内容になっているはずです.
structure{
atom_list{
atoms{
#tag element rx ry rz weight mobile
Si 0.9849 0 0.9166650000000001 1 off
Si 0.13255000000000006 0.2349 0.08333166666666668 1 off
Si 0.13254999999999992 0.7651 0.24999833333333335 1 off
...
...
mobile属性値は原子が「可動かどうか」を表すものですが,これがすべてoffという値に設定されています.分子動力学シミュレーションにおいてはすべての原子が可動であってほしいので,まずは一括置換などを用いてすべてonにしましょう.ついで,weight属性値が設定されていますが,この設定は不要です.そのかわりに原子が所属する熱浴を指定するthermo_group属性値は必要なので,weightをthermo_groupに書き換えましょう.幸い,その値はweightに対して設定してあった1という値で問題ありません.
さらにMDのステップ数やカットオフエネルギー,k点サンプリングなどの基本的な設定を行います.上で紹介したGitHubのレポジトリーでは以下のような設定が採用されています.
control{
condition = initial
cpumax = 47.5 hour
max_mdstep = 10000
max_iteration = 100000000
}
accuracy{
ksampling{
method = gamma
}
cutoff_wf = 25 rydberg
cutoff_cd = 225 rydberg
scf_convergence{
succession = 1
}
}
k点サンプリングは$\Gamma$点のみとしました.カットオフエネルギーは25 Rydbergです.MDステップ数は1万としています.
さらに,分子動力学シミュレーションの設定を行います.
structure_evolution{
method = temperature_control
dt = 1 fs
temperature_control{
method = temperature_control
num_thermostat = 1
set_initial_velocity = on
sw_shift_velocities = on
thermostat{
#tag temp
500
}
}
predictor{
sw_wf_predictor = on
}
nnp_output{
sw_nnp_output = on
filetype = xsf
frequency = 10
}
}
分子動力学シミュレーションに限らず,原子のダイナミクスの設定はstructure_evolutionブロックにおいて行います.ここでmethod=temperature_controlとすると温度一定の分子動力学シミュレーションを実行することができます.温度制御の詳細はtemperature_controlブロックで行います.いろいろな設定がほどこされていますが,重要なのはthermostatブロックにおいて設定されているtempの値です.これが温度に相当し,この例では500Kとなっています.ここの数値を変更すれば好きな温度のシミュレーションを行うことができます.nnp_outputブロックでは教師データ出力の設定を行っています.filetypeとしてxsfを指定していますが,これによってænetが理解できるxsf形式で原子座標や原子間力,エネルギーが出力されます.frequency=10としていますが,これによって10 MDステップに一度教師データが出力されることになります.
以上の設定がほどこされたPHASE/0のインプットファイルはGitHubのレポジトリーのSiO2/fpmd 以下のサブディレクトリーに配置されているのでご確認ください.
計算結果
PHASE/0を実行すると通常得られるファイル群のほか以下のようなファイルが得られます.
ls *.xsf
nfdynm000010.xsf nfdynm000020.xsf ...
これらが作りたかった教師データファイルです.GitHubのレポジトリーでは温度500K, 800K, 1200Kにおいてそれぞれ1万MDステップずつシミュレーションを行うような設定がほどこされていますが,10 MDステップに一度教師データをサンプルする設定がほどこされているので,完走すれば合計3000個の教師データが得られることになります.
結果のxsfファイルは対応する入力が存在するディレクトリーの下にbz2形式で圧縮して配置されています(ファイル名はxsf.tar.bz2)第一原理分子動力学シミュレーションをスキップしたい場合以下の要領で解凍してください.
tar -jxvf xsf.tar.bz2
機械学習ポテンシャルの作成
いよいよ機械学習ポテンシャルの作成を行っていきます.教師データを記述子に変換し,ニューラルネットワークにフィットします.さらに得られたポテンシャルを用いて第一原理計算の結果をどの程度再現するかを検証していきます.
教師データ変換
教師データにおける原子座標はカルテシアン座標で記述されています.これはそのままでは機械学習の記述子として用いることはできないので,変換処理を行います.ænetにおいてこの変換処理を担うのはgenerate.xプログラムです.対応する入力ファイルはGitHubのレポジトリーのSiO2/aenet/generate 以下に配置されています.
まずは,元素ごとに記述子を規定するためのfingerprint fileを作成します.以下のような内容になります.
ATOM Si
ENV 2
Si
O
RMIN 0.85d0
BASIS type=Chebyshev
radial_Rc = 6.0 radial_N = 16 angular_Rc = 6.0 angular_N = 8
BASISによって記述子の種類を選択します.Chebyshevという値を設定しています.これは分布関数をChebyshev多項式によって展開し,その展開係数を記述子として用いる手法です(参考文献 : N. Artrith, A. Urban, and G. Ceder, Phys. Rev. B 96 (2017) 014112) これがænetの推奨の方法のようです.次に重要なのが動径方向および角度方向のカットオフやメッシュ数です.radial_Rcで動径方向のカットオフを6.0 Åに,radial_Nで動径方向の分割数を16に,angular_Rcで角度方向のカットオフを6.0 Åに,angular_Nで動径方向の分割数を8に設定しています.このファイルを元素の数(今の場合2)だけ作成します.Si用のファイルがSi.fingerprint.stp, O用のファイルがO.fingerprint.stpです.
ついで,generate.xのコントロールファイルを作成します.以下のような内容になります.
OUTPUT SiO.train
TYPES
2
Si -107.171 | eV
O -434.109 | eV
SETUPS
Si Si.fingerprint.stp
O O.fingerprint.stp
FILES
3000
../../fpmd/800K/nfdynm000772.xsf
../../fpmd/800K/nfdynm000038.xsf
...
...
OUTPUTキーワードで変換結果を記録するファイルを指定します.SETUPSでは先に作成したfingerprint ファイルと元素名を紐づけます.FILESで教師データファイルのパスを指定します.続く行で教師データの数を設定し,さらに以降教師データファイル(PHASE/0が出力したxsf)のパスを指定します.SiO2/aenet/generateの下のgenerate.inがそのファイルです.
以上のファイルがそろったら,次の要領でgenerate.xを実行します.
$HOME/aenet-2.0.3/bin/generate.x generate.in > generate.out
結果はgenerate.inにおいて設定したSiO.train というファイルに記録されます.
ニューラルネットワークへのフィッティング
つぎに,教師データを用いてニューラルネットワークに対してフィッティングを行います.そのためのプログラムがtrain.xです.train.xの入力ファイルがGitHubのレポジトリーのSiO2/aenet/train/train.in です.その内容の主要な部分は下記の通り.
TRAININGSET SiO.train
TESTPERCENT 10
ITERATIONS 5000
METHOD
bfgs
NETWORKS
! atom network hidden
! types file-name layers nodes:activation
Si Si.10t-10t.nn 2 10:tanh 10:tanh
O O.10t-10t.nn 2 10:tanh 10:tanh
TRAININGSETで先の記述子に変換したファイルを指定します.カレントディレクトリーの下のSiO.train を指定しているので,genrate/SiO.trainファイルをtrainディレクトリーにコピーしましょう.TESTPERCENTで教師データの内テストにまわす割合を10%と設定しています.ITERATIONSではフィッティングの上限回数を5000回としています.METHODで最適化の手法としてbfgs法を採用することを指定しています.NETWORKS以下にフィットするニューラルネットワークを元素ごとに設定しています.
元素名 出力ファイル名 隠れ層の数 隠れ層毎のノード数:activation functionの種類
という形式で設定します.この例では隠れ層の数が2, 各隠れ層ごとのノード数は10でactivation functionとしてはhyperbolic tangentを用いるということを設定しています.以下の要領で実行します.
mpiexec -n 8 $HOME/aenet-2.0.3/bin/train.x train.in > train.out &
この処理は時間がかかるので,MPI並列を用いて実行しています.また,バックグランドで実行しています.train.outファイルには前処理の結果がいろいろと記録されたあと,以下の要領でフィッティングの結果が報告されます.
|------------TRAIN-----------| |------------TEST------------|
epoch MAE <RMSE> MAE <RMSE>
0 3.612219E-02 4.090257E-02 3.600668E-02 4.080010E-02 <
1 3.612219E-02 4.090257E-02 3.600668E-02 4.080010E-02 <
2 1.048382E-01 1.076145E-01 1.066514E-01 1.093361E-01 <
3 2.815346E-02 3.356589E-02 3.022437E-02 3.466527E-02 <
4 2.669718E-02 3.210864E-02 2.860923E-02 3.311245E-02 <
....
....
epochは最適化のステップ数,MAEはmean average error, <RMSE>はroot mean square error に対応します.単位はeV/atomです.それぞれ訓練データとテストデータにわけて出力されるようになっています.
生成されるポテンシャルファイルは,ステップごとに出力されます.
ls *.nn-*
O.10t-10t.nn-00001 ... Si.10t-10t.nn-00010 ...
...
元素名.ニューラルネットワークの設定.nn-ステップ数というファイル名で保存されます.ログを確認し,よくフィットできていると考えられるポテンシャルを採用しましょう.
ステップ数とRMSEの関係をプロットしたのが次の図です.あるところから特にテストデータに対するRMSEはあまり改善しなくなりますが,逆に悪くなるようなこともないようです.RMSEがおおよそ1 meV/atom程度までフィッティングができていることが分かります.
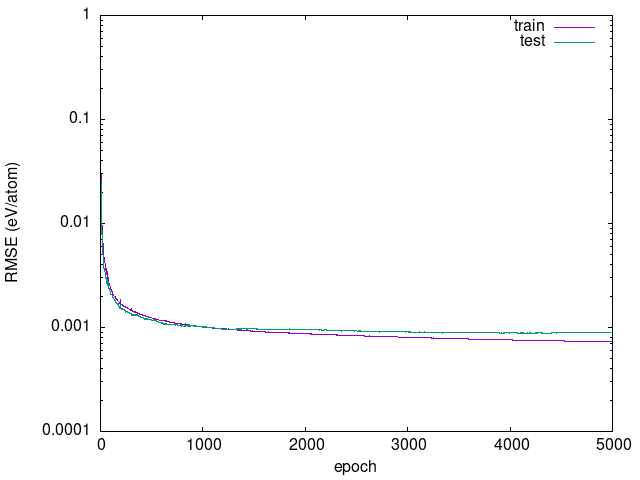
後半のステップならどれを採用しても大きな違いはないと考えられますが,目安としてテストデータに対するRMSEが最も小さいステップの結果を以降採用することにします.
検証
できたポテンシャルの検証をænetに付属するpredict.xプログラムを用いて行います.predict.x はxsfとポテンシャルファイルを入力とし,エネルギーや原子間力の計算および構造最適化を行うことができます(predict.xの機能は限定的なので,本格的な分子動力学シミュレーションを実行する場合はLAMMPSを用います)
SiO2/aenet/predict/predict.inにpredict.xの入力ファイルが配置されています.以下のような内容になっています.
TYPES
2
Si
O
NETWORKS
Si Si.10t-10t.nn
O O.10t-10t.nn
FORCES
FILES
3000
../../fpmd/800K/nfdynm000772.xsf
../../fpmd/800K/nfdynm000038.xsf
../../fpmd/800K/nfdynm000086.xsf
...
TYPESで原子種の指定を行っています.NETWORKSでポテンシャルファイルを指定しています.SiO2/aenet/trainディレクトリーからよかったステップのポテンシャルファイルをコピーし,Si.10t-10t.nn O.10t-10t.nn とリネームしましょう.FORCESと記述するとエネルギーだけでなく原子間力も計算してくれます.FILES以降入力としたい座標データが記録されたxsfファイルを指定します.この部分はgenerate.inと全く同じ記述になっています.
predict.xは以下の要領で実行することができます.
$HOME/aenet-2.0.3/bin/predict.x predict.in > predict.out
結果は標準出力に,以下のような形式で出力されます.
Structure number : 1
File name : ../../fpmd/800K/nfdynm000772.xsf
Number of atoms : 72
Number of species : 2
...
...
Cartesian atomic coordinates (input) and corresponding atomic forces:
x y z Fx Fy Fz
(Ang) (Ang) (Ang) (eV/Ang) (eV/Ang) (eV/Ang)
--------------------------------------------------------------------------------------
Si 9.649365 8.409237 9.994378 0.198728 0.398985 -1.409910
Si 1.351787 1.864069 0.963064 2.680675 2.235593 -0.187496
Si 1.296394 6.654412 2.411131 1.066620 0.626369 1.453671
O 8.774874 1.120195 9.374453 -0.053996 1.123514 -0.522405
...
...
Total energy : -23594.01883535 eV
Mean force (must be zero) : 0.000000 0.000000 0.000000
...
...
Structure numberで何番目の原子配置かが報告されます.File nameでは対象のxsfファイルのファイル名が報告されます.Number of atoms は対象の系の原子数,Number of speciesは対象の系の元素数です.Cartesian atomic coordinates (input) and corresponding atomic forces:以降原子座標と対応する原子間力がそれぞれÅおよびeV/Å 単位で記録されます.Total energyでエネルギーの計算結果が記録されます.このファイルからエネルギーの値を抽出し,xsfファイルに記録されている第一原理計算のエネルギーと比較するスクリプトがGitHubのレポジトリーのscripts/extract_energy.pyです.-nのあとにpredict.xのログを指定して,実行してみましょう.
../../../scripts/extract_energy.py -n predict.out
max diff : 0.005056026756960819 eV/atom, and the corresponding directory : ../../fpmd/1200K/nfdynm000783.xsf
average diff : 0.0006159564258358235 eV/atom
rmse : 0.0008024776361951128 eV/atom
max diffのあとに第一原理計算との誤差が最大だった系の情報が,average diffには平均誤差が,rmse のあとにRMSEが出力されます.また,energy.datファイルにニューラルネットワークポテンシャルのエネルギーと第一原理計算のエネルギーが記録されます.energy.datファイルの情報をプロットすると次のような図が得られます.
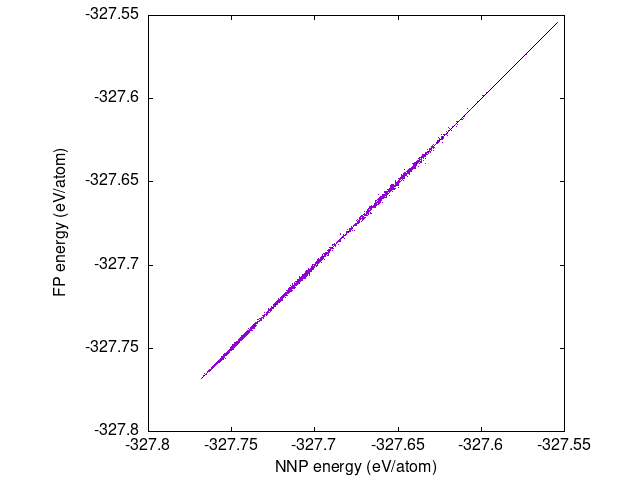
この図の各点は,完璧な学習ができていれば斜め45度の直線にのります.学習が完璧なことはないので直線から多少はばらついていますが,おおむね直線から大きくはずれてはいなく,うまく学習できたといえると思います.
LAMMPSを用いた分子動力学シミュレーション
先にコンパイルしたLAMMPSを用いて分子動力学シミュレーションを実行する方法を紹介します.LAMMPSの基本的な使い方についてはQiitaにも有用な記事が多数投稿されているので 詳しくない方はこれらを参照するとよいでしょう.
単位はmetal, atom_styleはatomicを用います.
units metal
atom_style atomic
ænetで作成したポテンシャルファイルを読み込むには,以下のような設定を施します.
pair_style aenet
pair_coeff * * v00 Si O 10t-10t.nn Si O
pair_styleにaenetを指定するとænetで作ったポテンシャルファイルを利用することができます.ポテンシャルファイルのファイル名はpair_coeff コマンドで指定します.はじめにアスタリスク二つを指定しすべての元素が対象であることを設定し,さらにv00によってænetが作成するファイルを用いることを指定します.つづくSi O 10t-10t.nnはSi.10t-10t.nn, O.10t-10t.nnの2つのファイル指定の短縮形で,最後のSi Oは元素へのマッピングで、1つ目のファイルはSi, 2つ目のファイルはOに対応する重みファイルであることを意味します.座標データファイルはatom_style=atomicの際に用いる形式で準備します.
500Kと800Kで分子動力学シミュレーションを実施してみました.対応するLAMMPSの入力スクリプトはSiO2/aenet/mdtest/の500Kおよび800Kに配置されています.初期原子配置としては第一原理分子動力学シミュレーションでも利用した原子配置を採用し,時間きざみ1 fsで20000ステップの分子動力学シミュレーションを実行する設定がほどこされています.各ディレクトリー直下にポテンシャルファイルが存在することが仮定されているので,実行前にポテンシャルファイルをコピーするのを忘れないようにしてください.
次の図に二体分布関数の計算結果を示します.教師データ作成に用いた第一原理分子動力学シミュレーションから得られた結果もあわせてプロットしています.
500Kの場合

800Kの場合

500K, 800K双方ともほとんど見分けがつかないレベルで第一原理計算と一致する結果が得られました.一方計算負荷ははるかに小さく,今回用いた系の場合おおよそ百分の一です.第一原理計算の計算時間は$O\left(N^3\right)$であるのに対しニューラルネットワークポテンシャルの計算時間は$O\left(N\right)$なので,系が大きくなればこの差は開く一方になります.
限界
ニューラルネットワークポテンシャルは対象の原子配置が教師データの補間として記述できる場合に威力を発揮しますが,補外になってしまうような原子配置に対しては全く機能しない場合があります.たとえば,1200Kで先の分子動力学シミュレーションを実行してみると,次のような二体分布関数が得られてしまいました.
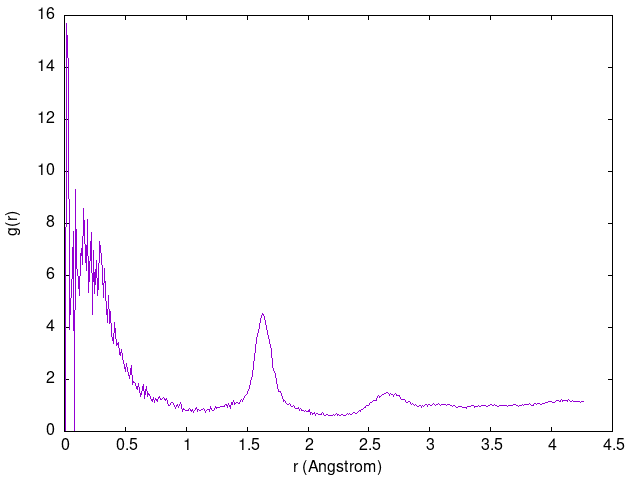
明らかにおかしな分布が短距離の領域で現れており,シミュレーションとしては破綻してしまったと言わざるを得ません.実利用の際はこの限界を意識し,シミュレーションを行いたい対象に応じて教師データを作成する必要があります.
おわりに
少し長くなってしまいましたが,機械学習の技術を用いて原子間ポテンシャルを作成する方法を紹介しました.必要なアプリケーション群のインストール方法,教師データの作り方,ニューラルネットワークポテンシャルの作り方,ニューラルネットワークポテンシャルを用いたシミュレーションの実行方法などについて説明しました.ここで行った説明の入力と出力(の一部)を公開しているので,興味があれば自分でも手を動かしてみてください.
また,ニューラルネットワークポテンシャルを作成するアプリケーションとしては,今回用いたænetのほかn2p2 やdeepmd などがあります.それぞれ定評のあるアプリケーションなので,興味があれば試してみてもよいのではないでしょうか.