Volcano plotとは
Volcano plotは、RNA-seqやマイクロアレイで二群の遺伝子発現量を比較する際に、遺伝子の発現比と統計的有意性(p値)でプロットした図です。
x軸を発現比、y軸を統計的有意性としたときのプロットが一般的な描き方です。
何千、何万という遺伝子の発現を視覚化して、重要な変化をしている遺伝子を特定することができます。
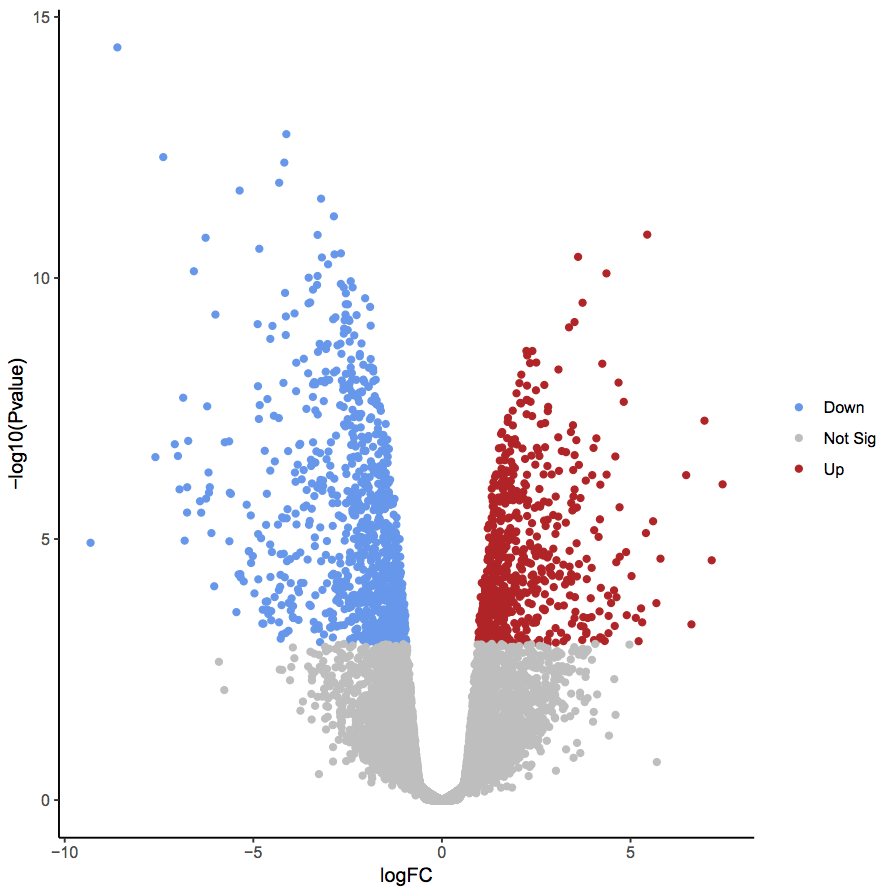
この図だと赤が統計的有意に発現量が増加した遺伝子で、青が統計的優位に発現量が減少した遺伝子を示しています。
エクセルでも作れますが、Pythonでbioinfokitライブラリを使えば、めっちゃ簡単に、しかもかっこいい図が作れちゃいます。
今回はこのVolcano plotの作り方を記していきます!
ライブラリのインストール
pipでもcondaでもインストールできますが、今回はcondaインストールをやっていきます。
下のコマンドを実行します。
conda install bioinfokit
これでbioinfokitライブラリはインストールできましたが、bioinfokitライブラリは他に色々なライブラリに依存しているので、入ってなければ必要なほかのライブラリ(adjusttextとかtextwrap3など)も入れていきます。
このあとfrom bioinfokit import analys, visuzを実行したときに、足りないものがあるとerrorになるので、errorとして出てきたものをインストールしていけば大丈夫です。
下のリンクの「Depends」を見れば必要なライブラリ一覧がわかります。
データの準備
必要なデータセットは、1) 遺伝子名、2) 比較したい二群(A、B)の遺伝子ごとのt検定の結果(p値)、3) 二群の発現比(A/B)です。つまり3カラム分です。
今回のデモデータは以下のURLのものを使います。
Volcano plotを描いてみる
早速描いていきます!
まずはライブラリのインポートです。使うライブラリはpandasとbioinfokitの二種類です。
import pandas as pd
from bioinfokit import analys, visuz
ここで、さっき言ったライブラリ(adjusttextとかtextwrap3など)が入っていないというエラーがあれば、condaインストールで入れていきます。
つぎにデータを読み込んで、中身を確認します。
df = pd.read_csv('testvolcano.csv')
print(df.head())
GeneNames log2FC p-value
0 LOC_Os09g01000.1 -1.886539 1.250000e-55
1 LOC_Os12g42876.1 3.231611 1.050000e-55
2 LOC_Os12g42884.2 3.179004 2.590000e-54
3 LOC_Os03g16920.1 5.290677 4.690000e-54
4 LOC_Os05g47540.4 4.096862 2.190000e-54
遺伝子名(GeneNames)、遺伝子の発現比(log2FC)、p値(p-value)の必要な3カラムを確認しました。
これでひとまず、必要なものは揃いました!
めっちゃ簡単です!
さっそく描いていきましょう!
引数は1) データフレーム名(df)、2) 発現比(log2FC)、3) p値(p-value)を与えるだけです。
visuz.GeneExpression.volcano(df=df, lfc='log2FC', pv='p-value')
そうすると、、、
こんな簡単に描けました!しかも、10行以内のコードで!
図は作業ディレクトリにPNG形式で保存されています。
緑色が統計的有意(p < 0.05)に発現が2倍以上増加した遺伝子群、赤色が統計的に有意に発現が2倍以上減少した遺伝子群です。
整えます
せっかくpythonで作ったのにこのままだと味気ないので、色々いじってみます。
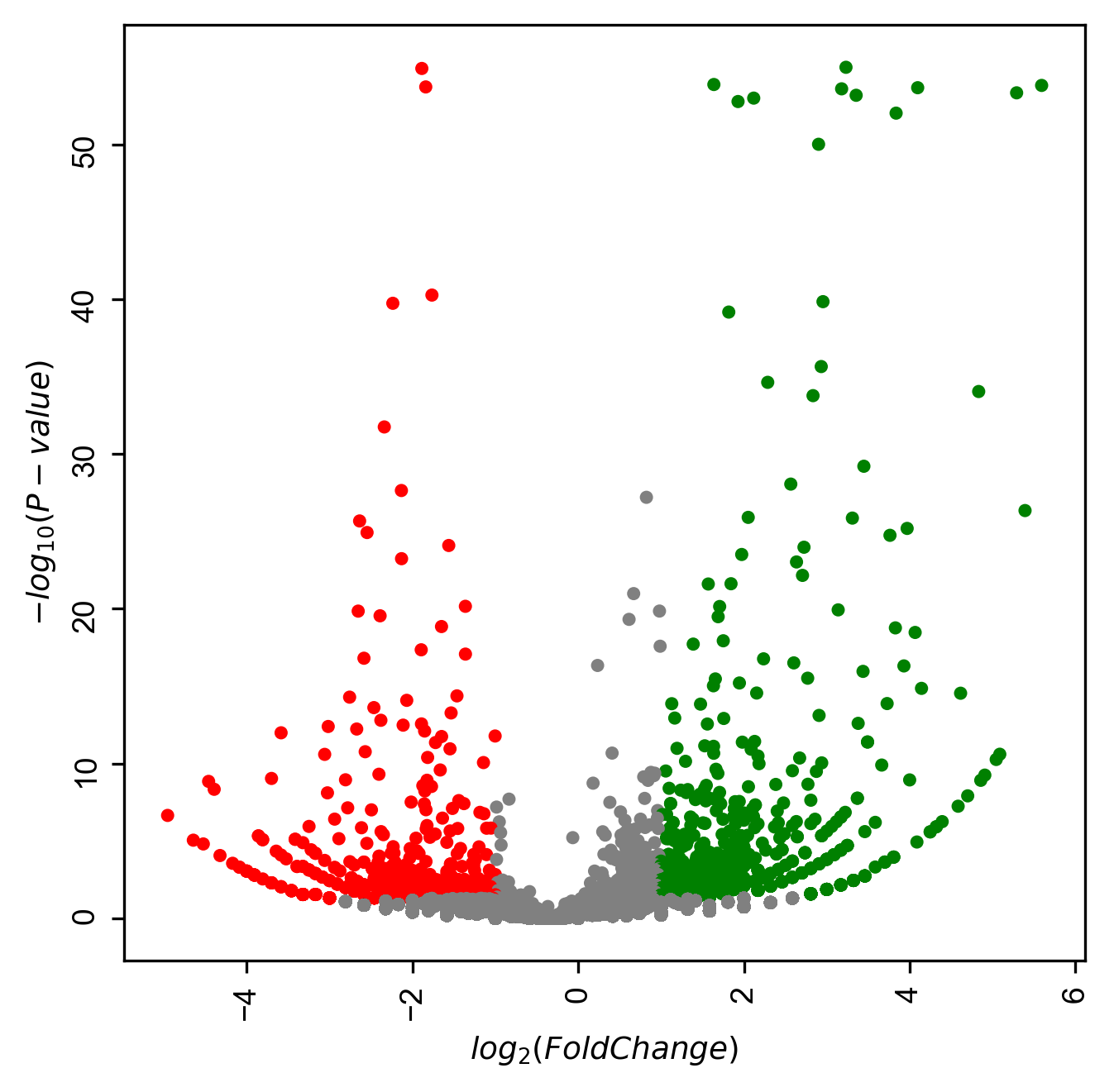
まずは色を変えてみます。発現が上がったらなんとなく赤色で、下がったらなんとなく青色な先入観があるので変えてみます。
visuz.GeneExpression.volcano(df=df, lfc='log2FC', pv='p-value',
color=('red', 'grey', 'blue')) #👈
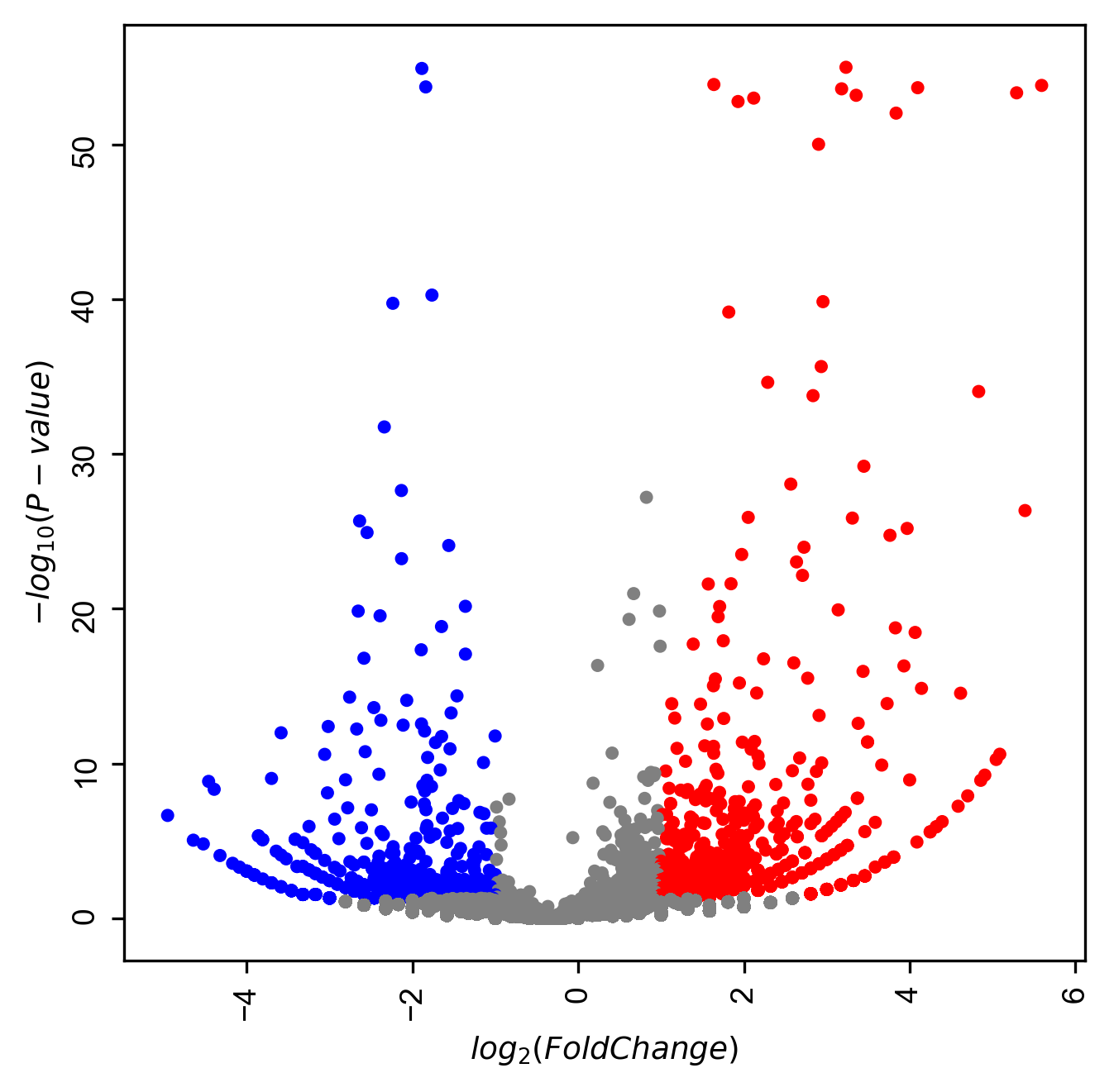
しきい値がどこらへんなのか、ラインがあるとわかりやすいですよね。しきい値ライン、入れてみます。
# ボルケーノプロット3
visuz.GeneExpression.volcano(df=df, lfc='log2FC', pv='p-value',
color=('red', 'grey', 'blue'),
sign_line=True) #👈
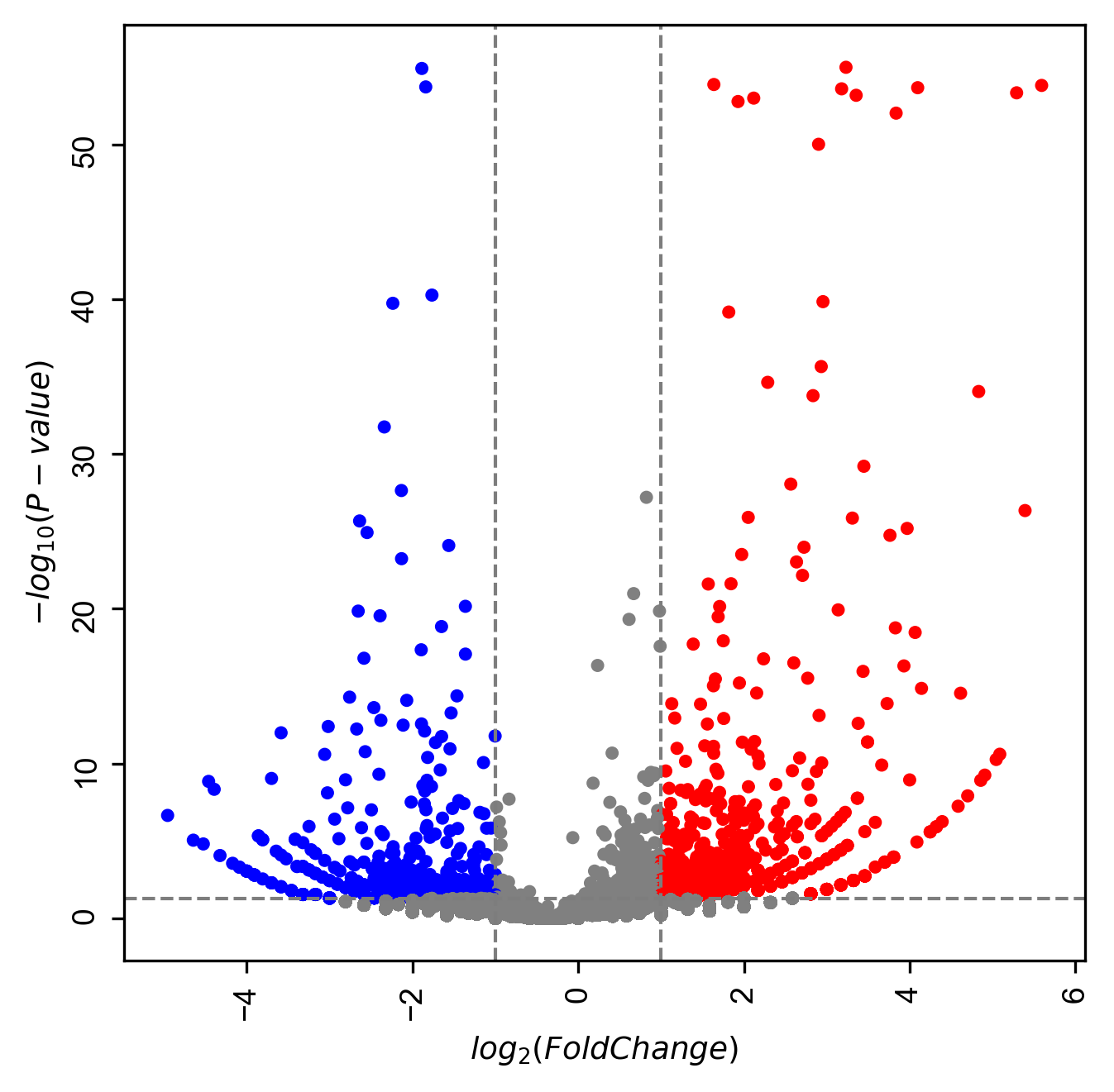
しきい値自体を変えることもできます。
デフォルトだと、低2の対数をとった発現比(log2FC)が1以上(つまり発現比に戻すと2倍以上)、p値が0.05未満になっています。
発現減少側(青色)のしきい値は変更せずに、発現増加側(赤色)のしきい値をlog2FCが2以上、p値が0.01未満としてみましょう。
visuz.GeneExpression.volcano(df=df, lfc='log2FC', pv='p-value',
color=('red', 'grey', 'blue'),
sign_line=True,
lfc_thr=(2, 1), #👈 発現比
pv_thr=(0.01, 0.05) #👈 p値
)
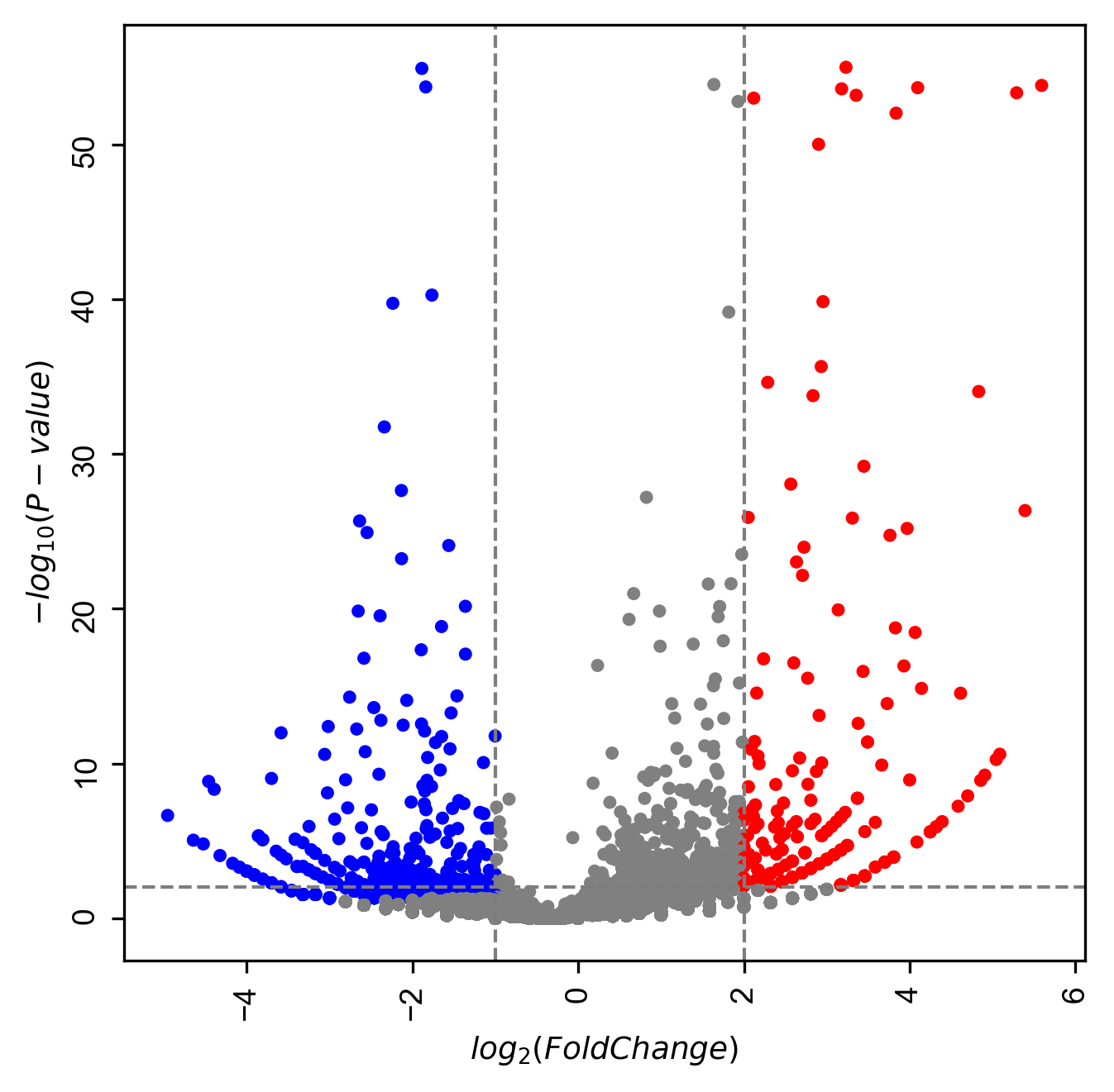
しきい値が二段階になっているので、ラインが機能していません。こういう場合は、ラインは消したほうが良さそうですね。
プロットに遺伝子名を付与していきます。引数geneidに遺伝子名のカラムを指定して、入れたい遺伝子名を引数genenamesで指定していきます。
visuz.GeneExpression.volcano(df=df, lfc='log2FC', pv='p-value', geneid='GeneNames', #👈
color=('red', 'grey', 'blue'),
sign_line=True,
genenames=('LOC_Os12g42876.1', 'LOC_Os09g01000.1', 'LOC_Os09g27030.2') #👈
)
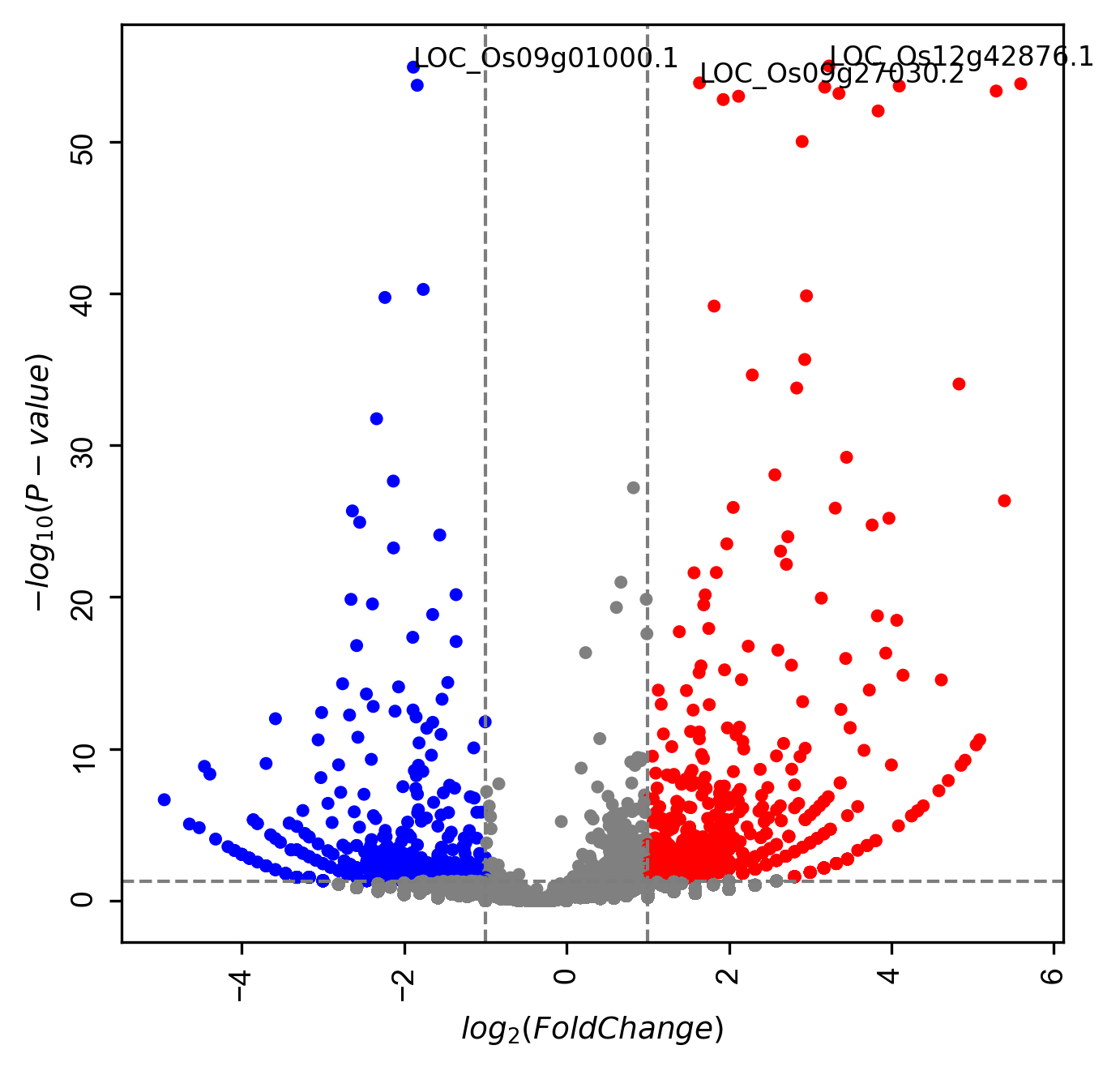
簡単にプロットと遺伝子名をリンクすることができました!
めっちゃ簡単です!
ちなみに、SpyderなどのIDEを使っていれば、引数にshow=Trueと入れれば、プロットのところに図が出力されます。
他にも色々ビジュアルを変えられるので試してみてください!