遺伝子発現ヒートマップとは
ヒートマップは、マイクロアレイやRNA-Seqの解析結果を視覚化する目的で使われます。全遺伝子のサンプル(実験区)ごとの発現の変化を視覚化するのに優れています。
遺伝子発現レベルに応じて色付けをしてサンプルごとの遺伝子発現レベルの変化を見ることができます。
このヒートマップにクラスタリング解析を組み合わせることで、よりグラフを見やすくすることができます。クラスタリング解析によって、遺伝子発現パターンが類似しているサンプル、遺伝子を見分けることができます。

(wikipediaより)
論文でもよく見かける何やら作るのが難しそうな図は、Pythonのbioinfokitライブラリを使えば簡単に作ることができちゃいます。
今回はこの遺伝子発現ヒートマップの作り方を備忘録的に書いていきます。
ライブラリのインストール
pipでもcondaでもインストールできますが、今回はcondaインストールをやっていきます。
下のコマンドを実行します。
conda install bioinfokit
これでbioinfokitライブラリはインストールできましたが、bioinfokitライブラリは他に色々なライブラリに依存しているので、入ってなければ必要なほかのライブラリ(adjusttextとかtextwrap3など)も入れていきます。
このあとfrom bioinfokit import analys, visuzを実行したときに、足りないものがあるとerrorになるので、errorとして出てきたものをインストールしていけば大丈夫です。
下のリンクの「Depends」を見れば必要なライブラリ一覧がわかります。
データの準備
使うデータは、インデックス(行)が遺伝子名で、カラム(列)が各条件の遺伝子発現量のテーブルデータです。
遺伝子発現ヒートマップを描いてみる
from bioinfokit import analys, visuz
import pandas as pd
ここで、さっき言ったライブラリ(adjusttextとかtextwrap3など)が入っていないというエラーがあれば、condaインストールで入れていきます。
つぎにデータを読み込んで、中身を確認します。
df = pd.read_csv('hm_cot.csv')
print(df.head())
Gene A B C D E F
0 B-CHI 4.505700 3.260360 -1.249400 8.89807 8.05955 -0.842803
1 CTL2 3.508560 1.660790 -1.856680 -2.57336 -1.37370 1.196000
2 B-CHI 2.160030 3.146520 0.982809 9.02430 6.05832 -2.967420
3 CTL2 1.884750 2.295690 0.408891 -3.91404 -2.28049 1.628820
4 CHIV 0.255193 -0.761204 -1.022350 3.65059 2.46525 -1.188140
遺伝子名をインデックスにします。
df = df.set_index(df.columns[0])
print(df.head())
A B C D E F
Gene
B-CHI 4.505700 3.260360 -1.249400 8.89807 8.05955 -0.842803
CTL2 3.508560 1.660790 -1.856680 -2.57336 -1.37370 1.196000
B-CHI 2.160030 3.146520 0.982809 9.02430 6.05832 -2.967420
CTL2 1.884750 2.295690 0.408891 -3.91404 -2.28049 1.628820
CHIV 0.255193 -0.761204 -1.022350 3.65059 2.46525 -1.188140
早速、さっそく描いていきます。
引数はデータフレームを与えるだけです。これで最低限のものは描けちゃいます。
visuz.gene_exp.hmap(df=df)
ワーキングディレクトリにpngファイルとして保存されています。

遺伝子名が重なってしまっているので、調整していきます。
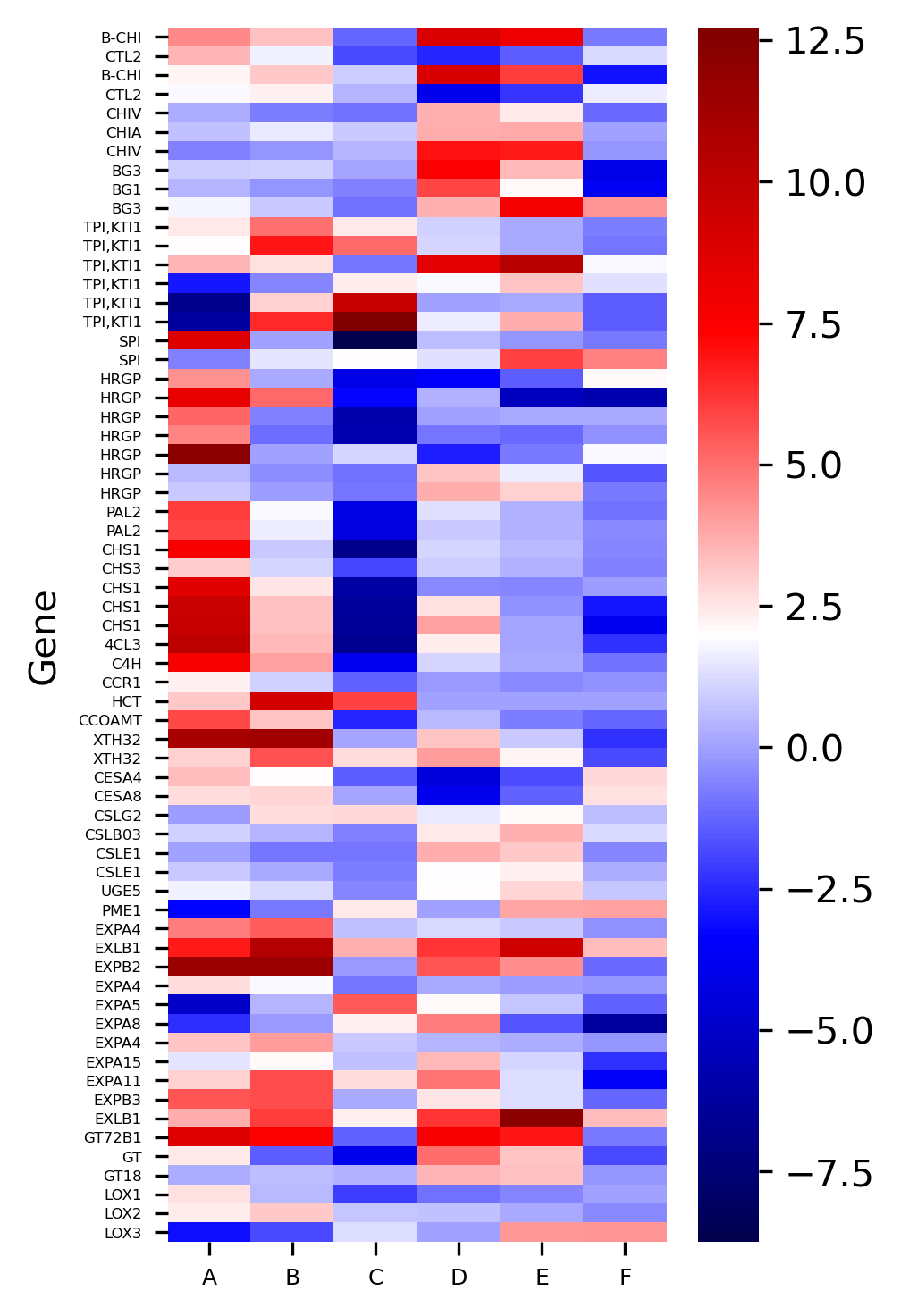
visuz.gene_exp.hmap(df=df, dim=(3, 6), tickfont=(6, 4), figname='heatmap1')
引数dimは図のサイズで(幅、高さ)を指定して、tickfontはX軸、Y軸のフォントサイズです。fignameは作成する図の名前を指定することができます。

階層クラスタリングを消すこともできます。行の階層クラスタリングを消す場合は引数rowclus=Falseを、列の階層クラスタリングを消す場合は引数colclus=Falseとします。
visuz.gene_exp.hmap(df=df, dim=(3, 6), tickfont=(6, 4), rowclus=False, colclus=False, figname='heatmap2')
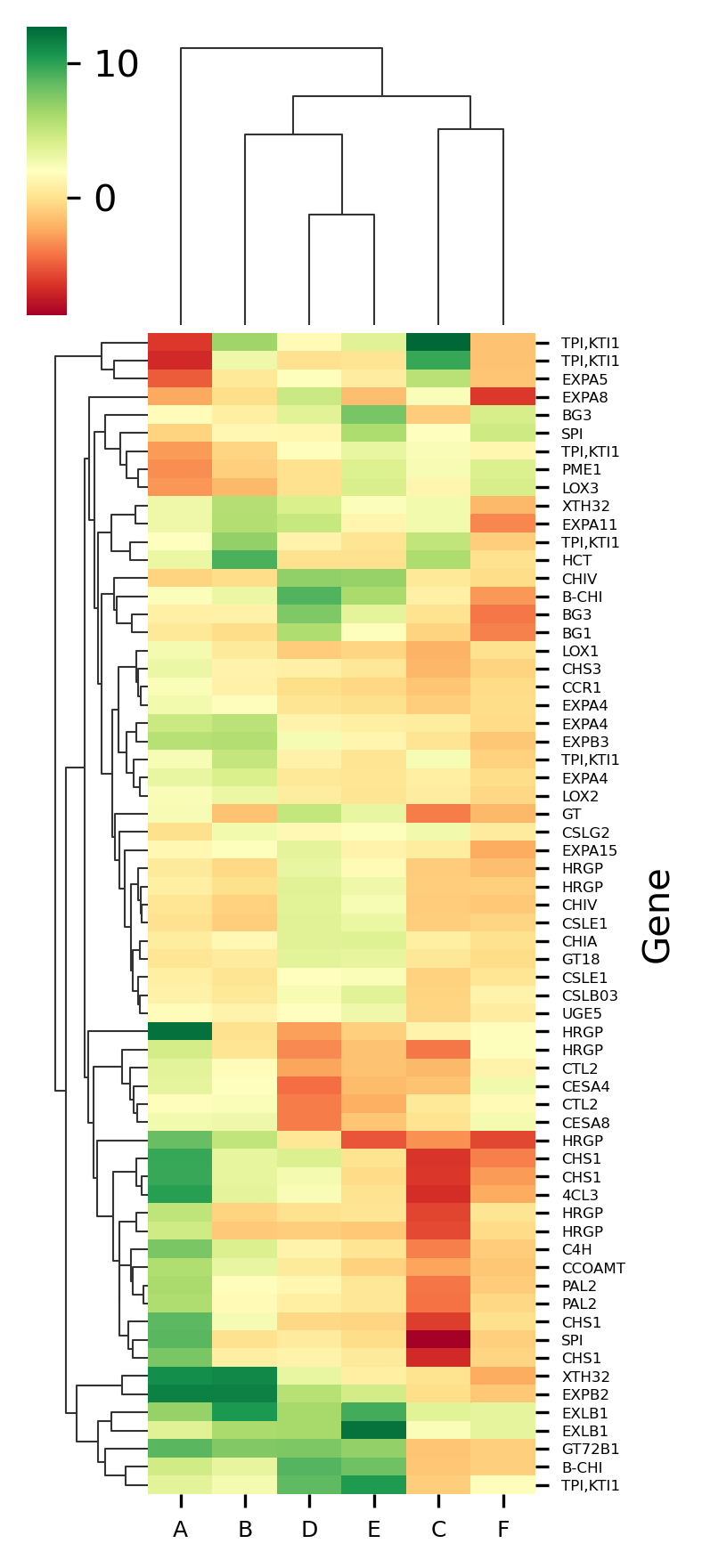
色を変えることもできます。引数cmapを変えていきます。デフォルトでは'seismic'となっていますが、'RdYlGn'とします。
visuz.gene_exp.hmap(df=df, dim=(3, 6), tickfont=(6, 4), figname='heatmap1')
さらに標準化した値でヒートマップを作成することもできます。
列を標準化したい場合はzscore=1、行を標準化したい場合はzscore=0とします。
visuz.gene_exp.hmap(df=df, dim=(3, 6), tickfont=(6, 4), zscore=0)

visuz.gene_exp.hmap(df=df, dim=(3, 6), tickfont=(6, 4), zscore=1)

こんな簡単にきれいな図が描けてしまいます。
他にも色々と引数を指定することで図を整えることができるのでやってみてください。