目次
・ 初めに
・ 概要
・ Step1.トポロジーを準備する
・ Step2.トポロジーを調べる
・ Step3.ユニットセルの定義と溶媒の追加
・ Step4.イオンの追加
・ Step5.エネルギー極小化
・ Step6.NVT平衡化
・ Step7.NPT平衡化
・ Step8.Production MD
・ Step9.解析
・ まとめ
・ 参考文献
初めに
- 勉強も兼ねてGROMACS Tutorial1を日本語化していきたいと思います。
- 単純に訳しただけです。
- 初学者のため、誤った認識をしている場合があります。間違いがあればご指摘の方お願いいたします。
- 使用しているのは2023-Homebrewです。
概要
この例ではイオンや水の箱の中のタンパク質(リゾチーム)を含むシミュレーション系を設定するプロセスについて説明します。各ステップで典型的な設定を用いたインプットとアウトプットの説明が含まれています。
Step1.トポロジーを準備する
構造の入手及び前処理
このチュートリアルではリゾチーム(PDB : 1AKI)を使用します。RCSBのウェブサイトにアクセスし、結晶構造のPDBファイルをダウンロードします。
ダウンロードした構造ファイルをVMD,Chimera,PyMOLで表示してみると結晶水があることがわかります。今回のチュートリアルでは結晶水は必要ないので以下のコマンドでPDBファイルから結晶水を除去します。
grep -v HOH 1aki.pdb > 1AKI_clean.pdb
注意
このような手順が常に適切であるとは限らないことに注意してください(例えば、強固に結合した、あるいは機能的な活性部位に存在する水分子の場合など)。
MISSINGというコメントで表示される項目は、結晶構造には存在しない原子または残基全体を示すので、常に.pdbファイルをチェックしてください。欠落した原子/残基は、他のソフトウェアパッケージを使用してモデル化する必要があります。
入力に必要なファイルの生成
結晶水を除去し、必要な原子が全て存在することが確認できたので、GROMACSの最初のモジュールであるpdb2gmxに入力する準備ができました。pdb2gmxの目的は、3つのファイルを生成することです。
- トポロジーファイル
- 位置拘束ファイル
- 前処理された構造ファイル
トポロジーファイルにはシミュレーション内で必要な非結合パラメータ(原子の種類と電荷)や結合パラメータ(結合、角度及び二面角)といった分子の情報が含まれます。
次のコマンドでpdb2gmxを実行します。
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
構造がpdb2gmxによって処理され、力場を選ぶように促されます。
Select the Force Field:
From '/usr/local/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
力場には、トポロジーに書き込まれる情報が入ります。これは非常に重要な選択なので各力場についてよく読み、自分の状況に最も適しているものを選ぶ必要があります。このチュートリアルでは、OPLS-AA/L力場を使用します。コマンドプロンプトで「15」と入力し、「Enter」を押してください。
pdb2gmxに渡すことのできるオプションは他にも多数存在します。よく使われているものは以下の通りです。
- -ignh:PDBファイルのH原子を無視します。
- -ter:N末端とC末端の電荷状態を相互作用で割り当てます。
- -inter:Glu、Asp、Lys、Arg、Hisの電荷状態をインタラクティブに割り当て、ジスルフィド結合に関与するCysを選択できる。
これで1AKI_processed.gro、topol.top及びposre.itpの3ファイルが生成されました。1AKI_processed.groは力場内で定義された全原子を含むGROMACSフォーマットの構造ファイルです(つまりタンパク質のアミノ酸にH原子が付加されています)。topol.top ファイルは系のトポロジーです(これについては後で詳しく説明します)。posre.itpファイルには、重原子の位置を制限するために使用される情報が含まれています(これについては後で詳しく説明します)。
Step2.トポロジーを調べる
topol.topファイルを調べると#include "oplsaa.ff/forcefield.itp"という記述があります。これはOPLS-AA力場のパラメータを呼び出しています。以降のすべてのパラメータがこの力場に由来していることを示しています。次に重要な行は [ moleculetype ]ディレクティブで、その下に次のようなものがあります。
; Name nrexcl
Protein_A 3
Protein_A "という名前は、PDBファイルでタンパク質がchain Aとラベル付けされたことに基づいて、分子名を定義しています。結合した近傍のために3つの除外があります。除外に関する詳細はGROMACSのマニュアルに記載されていますが、この情報についての議論はこのチュートリアルの範囲を超えています。
次の節では、タンパク質中の[atom]を定義します。情報はカラムとして表示されます:
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 1 LYS rtp LYSH q +2.0
1 opls_287 1 LYS N 1 -0.3 14.0067 ; qtot -0.3
2 opls_290 1 LYS H1 1 0.33 1.008 ; qtot 0.03
3 opls_290 1 LYS H2 1 0.33 1.008 ; qtot 0.36
4 opls_290 1 LYS H3 1 0.33 1.008 ; qtot 0.69
5 opls_293B 1 LYS CA 1 0.25 12.011 ; qtot 0.94
6 opls_140 1 LYS HA 1 0.06 1.008 ; qtot 1
この情報の解釈は以下の通りです:
- nr:原子の番号
- type:原子の種類
- resnr:アミノ酸残基番号
- residue:アミノ酸の残基名
- atom:原子名
- cgnr:電荷グループ番号(電荷グループとは、整数の電荷の単位を定義するもので、計算の高速化に役立つ。)
- charge:電荷
- mass:原子量
- typeB, chargeB, massB:自由エネルギー摂動に使用される(ここでは説明しない)
その後に続く節には、[bonds]、[pairs]、[angles]、[dihedrals]があります。これらの節のいくつかは自明です(結合、角、二面体)。これらのセクションに関連するパラメータと関数タイプは、GROMACSマニュアルの第5章に詳しく説明されています。特殊な1-4の相互作用は、「pair」(GROMACSマニュアル5.3.4)に含まれます。
ファイルの残りは、位置拘束から始まり、他のいくつかの有用/必要なトポロジーの定義が含まれます。posre.itpファイルはpdb2gmxによって生成されたもので、平衡化中に原子を所定の位置に保つための力の定数を定義しています(これについては後で詳しく説明します)。
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
これで "Protein_A "分子タイプの定義が終了しました。トポロジーファイルの残りの部分は、他の分子の定義とシステムレベルの記述に費やされます。次の分子タイプ(デフォルト)は溶媒で、この場合はSPC/E水です。水の他の典型的な選択肢は、SPC、TIP3P、TIP4Pです。pdb2gmxに"-water spce "を渡すことでこれを選択しました。しかし、これらのモデルのすべてがGROMACS内に存在するわけではないことに注意してください。
; Include water topology
#include "oplsaa.ff/spce.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
見てわかる通り、水も1000kJ mol-1 nm-2の力の定数(kpr)を用いて位置拘束が可能です。
イオンパラメータは以下の通りです。
; Include generic topology for ions
#include "oplsaa.ff/ions.itp"
最後に、システムレベルの定義があります。[ system ]ディレクティブは、シミュレーション中の出力ファイルに書き込まれるシステムの名前を与えます。[ molecules ]ディレクティブは、システム内のすべての分子をリストアップします。
[ system ]
; Name
LYSOZYME
[ molecules ]
; Compound #mols
Protein_A 1
[molecules]に関するいくつかの重要な注意事項
- リストアップされた分子の順番は、座標ファイル(この場合、.gro)の分子の順番と正確に一致しなければならない。
- リストアップされる名前は、残基名などではなく、各生物種の [ moleculetype ] 名と一致しなければならない。
これらの具体的な条件を満たさない場合、grompp(後述)から名前の不一致や分子が見つからないなどの致命的なエラーが出ます。
Step3.ユニットセルの定義と溶媒の追加
GROMACSのトポロジーに詳しくなったところで、系の構築の続きです。この例では、単純な水系をシミュレートすることにします。
ボックスの定義と溶媒の充填には2つのステップがあります:
-
editconfモジュールを使ってboxの範囲を定義する。 -
solvateモジュールを使って、boxに水を入れる。
ここで、ユニットセルをどのように扱うかの選択を迫られます。このチュートリアルでは、単純な立方体のboxをユニットセルとして使用します。周期境界条件やboxの種類に慣れてきたら、ひし形12面体を強くお勧めします。その体積は、同じ周期的距離の立方体箱の71%であり、加える水分子の数を節約することができます。
ユニットセルの定義
editconfを使用してboxを定義しましょう
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
上記のコマンドは、タンパク質をボックスの中央に配置し(-c)、ボックスの端から少なくとも1.0nmの位置に配置します(-d 1.0)。ボックスの種類は立方体(-bt cubic)と定義されています。ボックスの端までの距離は重要なパラメータです。溶質ボックスの距離を1.0nmとすると、タンパク質の2つの周期的なイメージの間に少なくとも2.0nmの距離が存在することになります。この距離は、シミュレーションで一般的に使用されるカットオフスキームのほとんどに十分対応できるものです。
溶媒の追加
boxの定義ができたので、その中に溶媒(水)を入れることができます。溶媒和は、solvateを使って実現します:
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
タンパク質の設定(-cp)は前のeditconfステップの出力に含まれており、溶媒の設定(-cs)は標準的なGROMACSのインストールに含まれているものです。出力は1AKI_solv.groと呼ばれ、solvateにトポロジーファイル名(topol.top)を伝え、修正できるようにしています。
solvateで行ったのは、追加した水分子の数を記録し、それをトポロジーに書き込んで、変更を反映させることです。他の(水以外の)溶媒を使用した場合、solvateはトポロジーにこれらの変更を加えないことに注意してください。
Step4.イオンの追加
これで、荷電したタンパク質を含む溶媒和した系ができました。生命は正味の電荷では存在しないので、私たちはシステムにイオンを加えなければならなりません。
GROMACSの中でイオンを追加するツールはgenionと呼ばれています。genionが行うのは、トポロジーを読み込んで、水分子をユーザーが指定したイオンと置き換えることです。このファイルはGROMACSのgromppモジュール(GROMACSプリプロセッサー)によって生成されます。gromppが行うのは、座標ファイルとトポロジー(分子を記述したもの)を処理して、原子レベルの入力(.tpr)を生成することです。tprファイルには、システム内のすべての原子のパラメータが含まれています。
gromppでtprファイルを生成するには、mdp(分子動力学パラメータファイル)という拡張子の入力ファイルが必要です。gromppはmdpファイルで指定されたパラメータと座標、トポロジー情報を組み合わせて、tprファイルを生成することができます。
mdpファイルは通常、エネルギー極小化やMDシミュレーションを実行するために使用されますが、今回は単にシステムの原子記述を生成するために使用されます。mdpファイルの例(今回使用するもの)は、こちらからダウンロードできます。
実際には、このステップで使用するmdpファイルには、任意の正当なパラメータの組み合わせが可能です。
tprファイルを以下のコマンドで作成して下さい:
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
これで、ions.tprに系の原子レベルの記述ができました。 このファイル(ions.tpr)をgenionに渡すことにします:
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
プロンプトが表示されたら、イオンを埋め込むためのグループ13「SOL」を選択します。タンパク質の一部をイオンに置き換えることは避けたい。
genionコマンドでは、入力として構造/状態ファイル(-s)を与え、出力として.groファイルを生成し(-o)、水分子の除去とイオンの添加を反映するためにトポロジーを処理し(-p)、プラスとマイナスのイオン名を定義し(それぞれ-pnameと-nname)、正しい数のマイナスイオン(この場合、タンパク質上の+8電荷を相殺するための8個のCl-イオン)を追加して、タンパク質上の純電荷を無効にすることに必要なだけイオン追加するようにgenionに対して伝えます。また、-neutralと-concオプションを組み合わせて指定することで、単に中和するだけでなく、指定した濃度のイオンを添加することもできます。これらのオプションの使用方法については、genionのマニュアルページを参照してください。
これで、[ molecules ]ディレクティブは次のようになります:
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10636
CL 8
ユニットセルの定義、溶媒・イオンの追加を行なった結果、以下のような構造が出来上がります。

Step5.エネルギー極小化
MDを始める前に、エネルギー極小化(EM)と呼ばれるプロセスを行うことで系の立体的な衝突や不適切な形状を緩和します。
EMのプロセスは、イオンの添加とよく似ています。今回もgromppを使って構造、トポロジー、シミュレーションパラメータを入力ファイル(.tpr)にまとめますが、今回は.tprをgenionに渡すのではなく、mdrunでエネルギー極小化を実行します。入力パラメータであるminim.mdpはこちらからダウンロードできます。
gmx grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
-vはステップごとに進捗状況をスクリーンに表示します。-deffnmは入力と出力のファイル名を定義します。今回の場合、以下のようなファイルが得られます:
- em.log: EMのログファイル
- em.edr: エネルギーファイル(バイナリ)
- em.trr: トラジェクトリファイル(バイナリ)
- em.gro: エネルギー極小化された構造ファイル
EMが成功したかどうかを判断するために、評価すべき非常に重要な要素が2つあります。
1つ目は、ポテンシャルエネルギー(Epot)です。Epotは水中の単純なタンパク質の場合、負であるべきです。系のサイズや水分子の数にもよりますが、105~106のオーダーになります。
2つ目の重要な特徴は最大力Fmaxですminim.mdpでは「emtol = 1000.0」と設定されており、Fmaxが1000kJ mol-1 nm-1を超えないことが目標であることが示されています。
Fmax > emtolで妥当なEpotに到達することもあり得ます。このような場合、系はシミュレーションに十分な安定性を持っていない可能性があります。なぜこのようなことが起こるのかを評価し、極小化パラメータ(integrator、emstepなど)を変更する必要があるかもしれません。
ポテンシャル解析
em.edrファイルには、GROMACSがEM中に収集したエネルギー項がすべて含まれています。GROMACSのエネルギーモジュールを使えば、どの.edrファイルでも解析することができます:
gmx energy -f em.edr -o potential.xvg
プロンプトで「10 0」と入力すると、ポテンシャル(10)が選択され、ゼロ(0)で入力が終了します。Epotの平均値が表示され、"potential.xvg "というファイルが書き込まれます。このデータをプロットするには、Xmgraceプロットツールが必要です。Epotの収束が順調であることを示す、このようなプロットが出来上がるはずです:

さて、系のエネルギーが最小になったところで、ダイナミクスを開始します。
Step6.NVT平衡化
EMは、形状や溶媒の方向性という点で、妥当な開始構造を確保することができました。実際のダイナミクスを開始するには、タンパク質の周りの溶媒とイオンを平衡化させる必要があります。溶媒はそれ自身の中で最適化されていることがほとんどであり、必ずしも溶質と最適化されているわけではないため、この時点でダイナミクスを試みると、系が崩壊する可能性があります。従って、溶媒をシミュレーションしたい温度にして、溶質(タンパク質)に対して適切な向きを確立する必要があります。適切な温度(運動エネルギーに基づく)に到達したら、適切な密度になるまで系に圧力をかけます。
平衡化は、しばしば2つのフェーズで実施されます。第一段階は、NVTアンサンブル(粒子数、体積、温度が一定)の下で行われます。通常、50~100psで十分であり、本演習では100psのNVT平衡化を実施することにします。マシンによっては、しばらく時間がかかるかもしれません(16コアで並列実行した場合、1時間弱)。mdpファイルはこちらで入手してください。
EMステップの時と同じようにgromppとmdrunを呼び出します:
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
mdpファイルには以下の記述が含まれます。
- gen_vel = yes: 速度生成を開始します。異なるランダムシード(gen_seed)を使用すると、異なる初期速度が得られるので、同じ開始構造から複数の(異なる)シミュレーションを行うことができます。
- tcoupl = V-rescale: The velocity rescaling thermostat is an improvement upon the Berendsen weak coupling method, which did not reproduce a correct kinetic ensemble.
- pcoupl = no: 圧力カップリングは適用されません。
温度解析
energyを使って、温度進行を解析してみましょう:
gmx energy -f nvt.edr -o temperature.xvg
プロンプトで「16 0」と入力し、システムの温度を選択して終了します。出来上がったプロットは次のようなものになるはずです:

Step7.NPT平衡化
NVT平衡化では、システムの温度を安定させました。データ収集の前に、システムの圧力(したがって密度も)を安定させる必要があります。圧力の平衡化は、粒子数、圧力、温度がすべて一定であるNPTアンサンブルの下で行われます。このアンサンブルは「等温・等圧」アンサンブルとも呼ばれ、実験条件に最も近いとされています。
100psのNPT平衡に使用される.mdpファイルは、ここで見つけることができます。NVTの平衡化に使用したパラメータファイルと大きく異なるところはありません。Parrinello-Rahman barostatを使用した圧力カップリングセクションが追加されていることに注意してください。
- continuation = yes: NVTの平衡化段階からシミュレーションを継続する
- gen_vel = no: 軌跡から速度が読み込まれる(後述)
NVT平衡の時と同じように、gromppとmdrunを呼び出します。NVT平衡のチェックポイントファイルを含めるために、-tフラグを付けていることに注意してください。このファイルには、シミュレーションを継続するために必要なすべての状態変数が含まれています。NVT中に生成された速度を保存するために、このファイルを含める必要があります。座標ファイル(-c)は、NVTシミュレーションの最終出力です。
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx mdrun -deffnm npt
圧力解析
energyで圧力の進行を解析してみましょう:
gmx energy -f npt.edr -o pressure.xvg
プロンプトで「18 0」と入力し、システムの圧力を選択して終了します。プロットは次のようなものになるはずです:
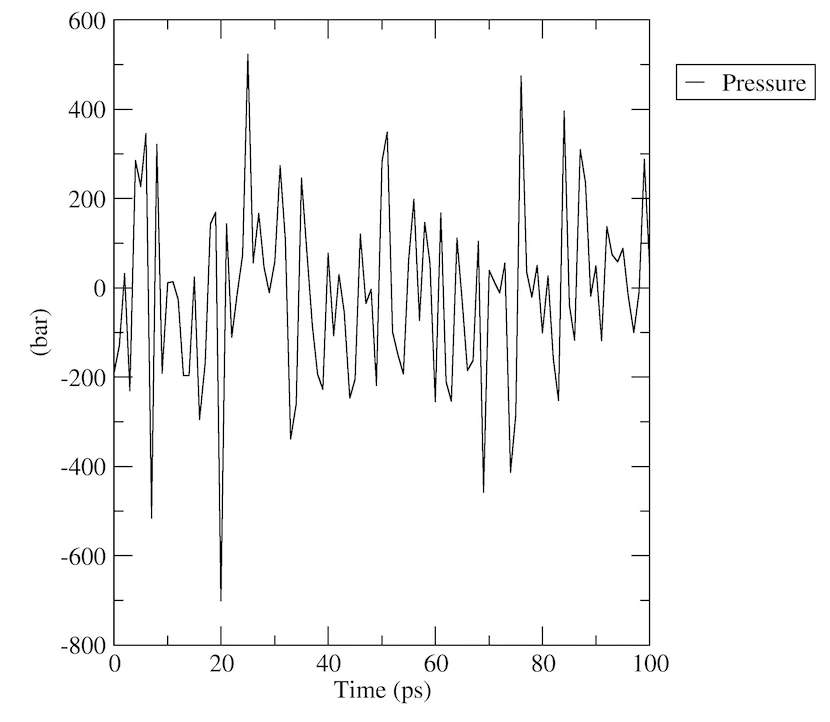
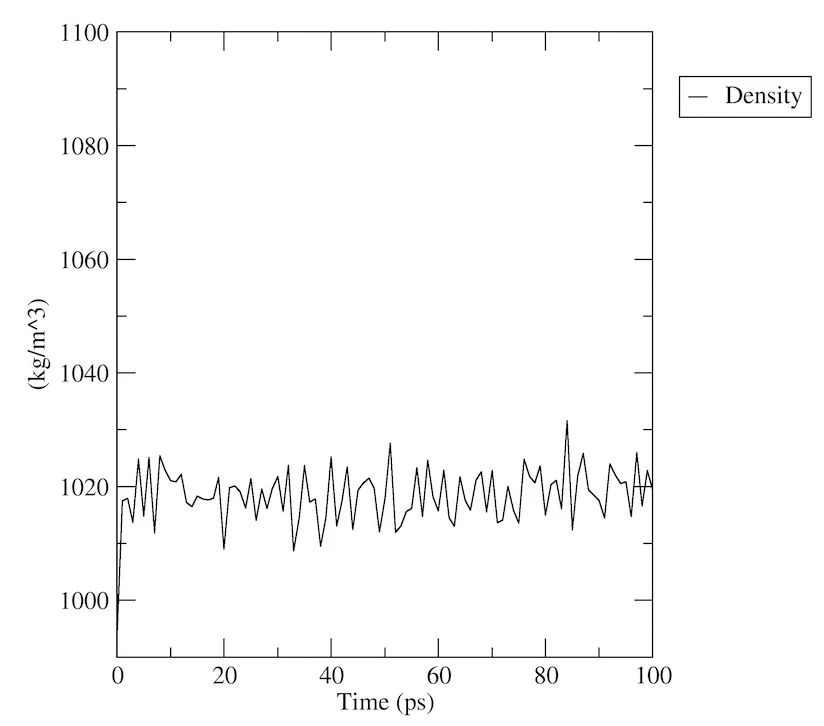
密度解析
今度はenergyを使って、プロンプトに「24 0」と入力して、密度も見てみましょう。
gmx energy -f npt.edr -o density.xvg
Step8.Production MD
2回の平衡化が完了すると、系は目的の温度と圧力で十分に平衡化されます。これで、位置拘束を解除し、プロダクションMDを実行する準備が整いました。以前のプロセスの通り、gromppのためにチェックポイントファイルを利用します。1nsのMDシミュレーションを実行しますが、そのmdpファイルはこちらにあります。
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
gromppはPME負荷の推定値を表示し、PMEやPP計算に何台のプロセッサを使用すべきかを決定します。詳細はGROMACS 4の出版物とマニュアルを参照してください。
立方体の場合、最適なPME負荷は0.25 (3:1 PP:PME)、12面体の場合、最適なPME負荷は0.33 (2:1 PP:PME)です。
mdrunを実行すると、プログラムは自動的にPPとPME計算の最適なプロセッサの数を決定します。したがって、計算のために適切なスレッド/コア数(-nt Xの値)を示し、最高のパフォーマンスを得ることができるようにしてください。
mdrunを実行してください:
gmx mdrun -deffnm md_0_1
GPUが利用可能な場合以下のコマンドが利用できます:
gmx mdrun -deffnm md_0_1 -nb gpu
Step9.解析
さて、タンパク質のシミュレーションができたので、システムの解析を実行しましょう。どのような種類のデータが重要なのでしょうか?これはシミュレーションを実行する前に問うべき重要な質問です。自分のシステムで収集したいデータの種類について、ある程度の考えを持つべきです。このチュートリアルでは、いくつかの基本的なツールを紹介します。
1つ目はtrjconvで、座標の除去、周期性の補正、軌道の手動変更(時間単位、フレーム周波数など)を行う後処理ツールとして使用されます。この演習では、trjconvを使ってシステムの周期性を考慮することにします。タンパク質はユニットセル内を拡散し、壊れているように見えたり、boxの反対側に飛び移るように見えたりすることがあります。そのような挙動を考慮するために、次のコマンドを実行してください:
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
センタリングするグループとして1("Protein")を選択し、出力には0("System")を選択します。この「補正された」軌跡ですべての解析を行うことになります。まず、構造安定性について見てみましょう。GROMACSにはrmsというRMSD計算のためのコマンドが組み込まれています。rmsを使うには、次のコマンドを実行します:
gmx rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg -tu ns
最小二乗法によるフィットとRMSD計算のグループの両方に4("Backbone")を選択します。-tuフラグは、軌跡がpsで書かれているにもかかわらず、結果をnsで出力します。これは、出力をわかりやすくするためです。出力プロットは、極小、平衡化された系に存在する構造に対するRMSDを表示します。
プロットしてみると、次のような結果になりました:

RMSDが0.1 nm (1 Å)付近であり、この構造が非常に安定していることを示しています。
タンパク質の回転半径は、そのコンパクトさの指標となります。タンパク質が安定に折り畳まれていれば、比較的安定したRg値を維持することができます。シミュレーションでリゾチームの回転半径を解析してみましょう:
gmx gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
解析のためにgroup 1 (Protein)を選択して下さい。

まとめ
GROMACSを使ったMDシミュレーションを行い、その結果の一部を解析しました。このチュートリアルは包括的なものではないと考えるべきです。GROMACSで実施できるシミュレーションは他にもたくさんあります(自由エネルギー計算、非平衡MD、normal modes analysisなど)。また、文献やGROMACSのマニュアルを見て、効率や精度のためにここで提供されているmdpファイルを調整する必要があります。
参考文献
この記事は以下の情報を参考にして執筆しました。