更新: バージョンが4.0.2に更新されています。If you ran analyses on eukaryotes using v4.0.0 and v4.0.1, please validate you result with v4.0.2 or higher, as the score may have been underestimatedとのこと。注意ください。
BUSCO
公式サイト: https://busco.ezlab.org/
BUSCOはCore gene setがアセンブルされた配列の中にどれだけあるか調べることで、ゲノムシーケンスやトランスクリプトームシーケンスがどれくらいの精度でできているかを調べるものである。例えば、とある菌にはCore gene setが400個あったとして実際には200個しか見つからなかったとしたら、「シーケンス段階としてはまだまだだね」という話になる。逆に99%以上あるとすれば、なかなか信頼できるシーケンスだなという一つの指標として利用できる。
Based on evolutionarily-informed expectations of gene content of near-universal single-copy orthologs, BUSCO metric is complementary to technical metrics like N50.
Citation:
Seppey M., Manni M., Zdobnov E.M. (2019) BUSCO: Assessing Genome Assembly and Annotation Completeness. In: Kollmar M. (eds) Gene Prediction. Methods in Molecular Biology, vol 1962. Humana, New York, NY. 2019 doi.org/10.1007/978-1-4939-9173-0_14. PMID:31020564
BUSCOはこれまでVersion3で稼働してきたが、Version4が先日リリースされた。当方は卒論生で、ゲノム情報から実験に繋がるようなデータを探している。最終目的がコンプリートゲノムを作ることではないので、今あるドラフトゲノムが十分信頼できるかということを示したいと思い、BUSCOを利用することにした。Bandageなどで確認しても、十分読めていると結論づけていたが、念のための確認したかった。
BUSCOに関してインターネットでざっと探したところ、多くの情報がVersion3までの情報であった。まだまだVersion3も利用できるが、やはり最新バージョンでやりたいというのが気持ちではある。実際最新バージョンでやろうとしたところ、少しトラブったので紹介していきたいと思う。
インストール
当方はいつものようにminicondaでインストールした。Docker等も利用できる。https://busco.ezlab.org/に他の方法が載っている。
conda install -c bioconda -c conda-forge busco=4.0.2
conda activate base
使い方
今までのBUSCOだとBUSCOとデータベースは別々にダウンロードするとのことだったが、バージョン4ではBUSCOが自動的に必要なデータベースを落としてくれるので、その必要がなくなった。なお、利用可能なデータベースは以下のコマンドで確認可能。BUSCOではデータベースではなくLineageという単語が用いられている。
busco --list-datasets
当方はlactobacillales_odb10が該当したので、これを利用する。基本的な利用方法は以下のコマンドの通りになる。
busco -m MODE -i INPUT -o OUTPUT -l LINEAGE
詳しい説明
-i or --input defines the input file to analyse which is either a nucleotide fasta file or a protein fasta file, depending on the BUSCO mode.
-o or --output defines the folder that will contain all results, logs, and intermediate data
-l or --lineage_dataset
It can be a dataset name, i.e. bacteria_odb10, or a path i.e. ./bacteria_odb10 or /home/user/bacteria_odb10. In the former case, which is the recommended usage, BUSCO will automatically download and version the corresponding dataset. In the latter case, the dataset found in the given path will be used. Lineage can be ignored if running automated lineage selection
-m or --mode sets the assessment MODE: genome, proteins, transcriptome
当方はゲノムモード、なおかつLineageはlactobacillales_odb10でやるので、
busco -m genome -i INPUT.nucleotides -o OUTPUT -l lactobacillales_odb10
でやることになった。(Input.nucleotides=fastaファイルでOK) なお、DependenciesとしてゲノムモードならtBLASTn, Prodigal (for non-eukaryotes) or Augustus (for eukaryotes), HMMERがあるので注意。
トラブル発生
早速コマンドを打ってやってみたが、普段見ないエラーが出てきた。ノートを取るのを忘れたので、正確なエラー名は分からないが
OSError: [Errno 24] Too many open files
と出てきた。調べていくと、どうやら現在開いているファイル数が、ファイルディスクリプタの上限に達してしまっていることが原因らしい。Qiitaでこのトラブルについて説明している人がいたので、この通りにやってみた。
当方のパソコンのスクリプトエディタの上限は256だった。
ulimit -n
256
そこで、これを
$ ulimit -n
256
$ ulimit -n 8192
unlimit -n
8192
とした。
その後、再度
busco -m genome -i hoge.fasta -o OUTPUT -l lactobacillales_odb10
を動かすと、問題なく動き、最後までデータが出力された。
結果の解釈
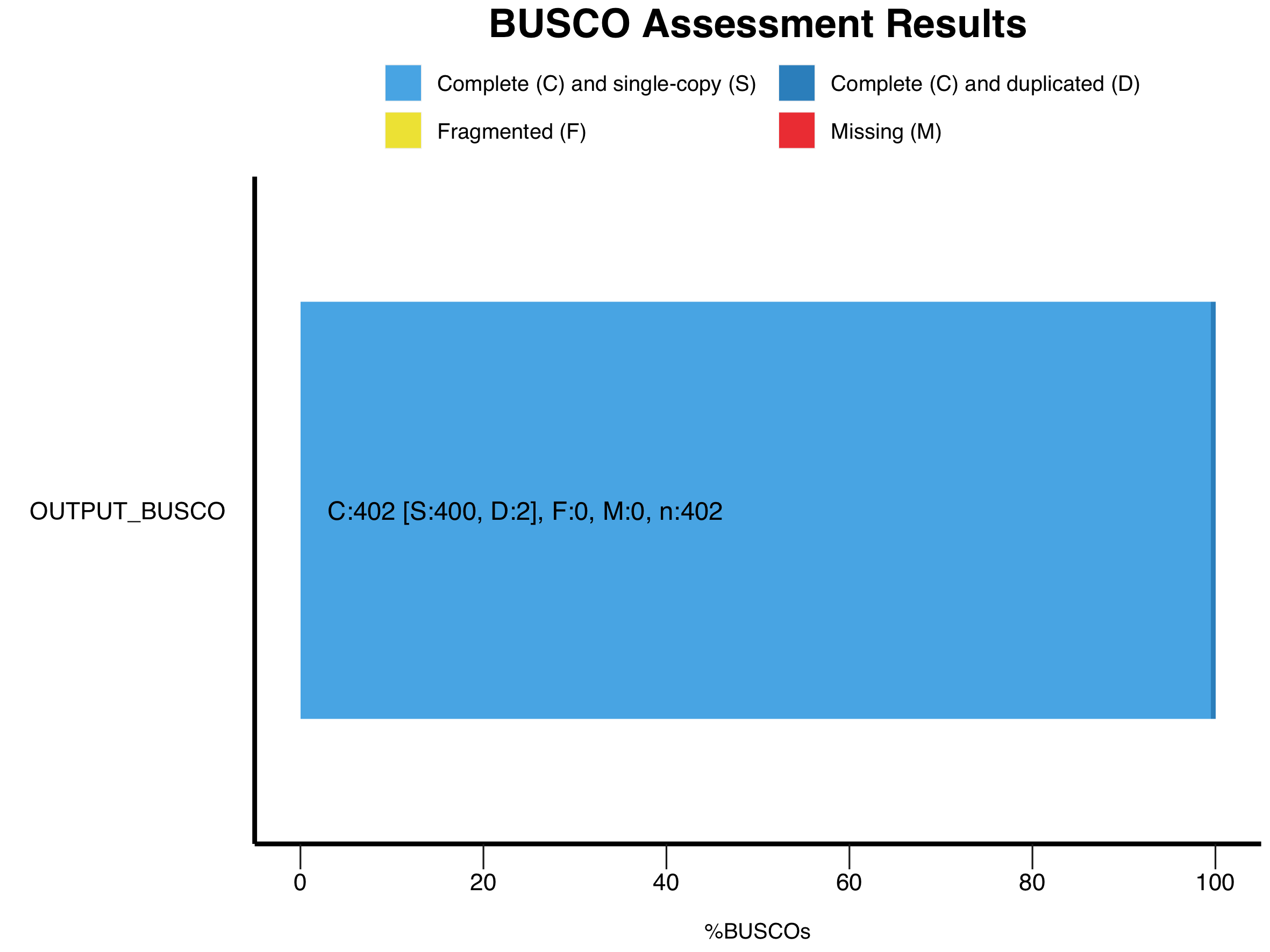
OUTPUTとして出てくるデータはいくつかあるが、重要なのはいくつのCore Gene Setが含まれていたかということである。当方の場合は100%だったので、問題なく解析を進められそうである。大量にデータを解析した場合は、generate_plot.pyを落とすといい。出力されてくるshort_summary.*.lineage_odb10.XX1.txtを一つのフォルダにまとめる。BUSCO_summariesといyディレクトリを作るとわかりやすいだろう。
mkdir BUSCO_summries
cp XX1/short_summary.*.lineage_odb10.XX1.txt BUSCO_summaries/.
cp XX2/short_summary.*.lineage_odb10.XX2.txt BUSCO_summaries/.
cp XX3/short_summary.*.lineage_odb10.XX3.txt BUSCO_summaries/.
なぜか、-wdではなく--working_directory PATHにしないと動かなかった。1番が公式の説明、2番が当方が動かせたコマンド。1番だと、うまくワーキングディレクトリとして動いてくれずにエラーが出た。
1. python3 scripts/generate_plot.py –wd BUSCO_summaries
2. python3 scripts/generate_plot.py --working_directory BUSCO_summaries