はじめに
仕事で統計遺伝学が必要になりそうなので、GWAS結果からマンハッタンプロットを描いてみた。
備忘録的なやつ。
使用するデータ
NIHのGRASPデータベースに様々なGWAS結果が落ちている。
そのうちの一つを使用してみる。
[Yang et al.] (https://grasp.nhlbi.nih.gov/downloads/ResultsOctober2016/Yang_Hypertension/Yang_YoungOnsetHypertension_Illumina550_SBAS_rawpv.csv)
必要なパッケージ
qqmanパッケージとdata.tableパッケージをインストールしておく。
qqmanパッケージはマンハッタンプロットの作図に必要。
GWAS結果は一般にファイルサイズが大きいため、高速にデータを読み込むため**fread()**関数を使用する。
fread()関数はdata.tableパッケージに入っている。
library(qqman)
library(data.table)
データの読み込み
**fread()**関数でデータを読み込む。
DF <- fread("Yang_YoungOnsetHypertension_Illumina550_SBAS_rawpv.csv",
stringsAsFactors = F, header = T)
**str()**関数でデータの内容を確認する。
実行結果。
> DF <- fread("Yang_YoungOnsetHypertension_Illumina550_SBAS_rawpv.csv",
+ stringsAsFactors = F, header = T)
> str(DF)
Classes ‘data.table’ and 'data.frame': 475157 obs. of 8 variables:
$ dbSNP_RS_ID : chr "rs3094315" "rs12562034" "rs3934834" "rs9442372" ...
$ Chromosome : int 1 1 1 1 1 1 1 1 1 1 ...
$ Physical_position: int 742429 758311 995669 1008567 1011278 1020428 1021403 1038818 1039813 1051029 ...
$ Symbol : chr "FLJ22639" "FLJ22639" "rs3934834" "C1orf159" ...
$ CLR_N_pv : num 0.977 0.706 0.651 0.673 0.731 ...
$ CLR_N_BMI_pv : num 0.357 0.298 0.348 0.844 0.429 ...
$ CLR_C_pv : num 0.692 0.438 0.513 1 0.527 ...
$ CLR_C_BMI_pv : num 0.114 0.0697 0.255 0.8723 0.2363 ...
- attr(*, ".internal.selfref")=<externalptr>
dbSNP_RS_ID:SNPs ID
Chromosome:染色体番号
Physical_position:染色体位置
CLR_N_pv、CLR_N_BMI_pv、CLR_C_pv、CLR_C_BMI_pv:p値(BMI調整あり/なし、条件付きロジスティック回帰)らしい...
マンハッタンプロットの描画
いよいよ描画。
qqmanパッケージの**manhattan()**関数を使う。
# マンハッタンプロットの描画
manhattan(DF, chr = "Chromosome",
bp = "Physical_position",
p = "CLR_C_BMI_pv",
snp = "dbSNP_RS_ID")
引数の説明。
chrには染色体番号のカラムを指定する。 X、Y、またはMT染色体がある場合は、これらの23、24、25などの番号に置換する。デフォルトはPLINKの「CHR」。
bpには染色体位置のカラムを指定する。デフォルトはPLINKの「BP」。この列は数値でなきゃダメ。
pはp値のカラム。デフォルトはPLINKの「P」。この列ももちろん数値でなきゃダメ。
※今回は「dbSNP_RS_ID」とした。
snpはSNP名のカラム(rs番号)を指定する。デフォルトはPLINKの「SNP」。このカラムは文字列でなきゃダメ。
描画結果
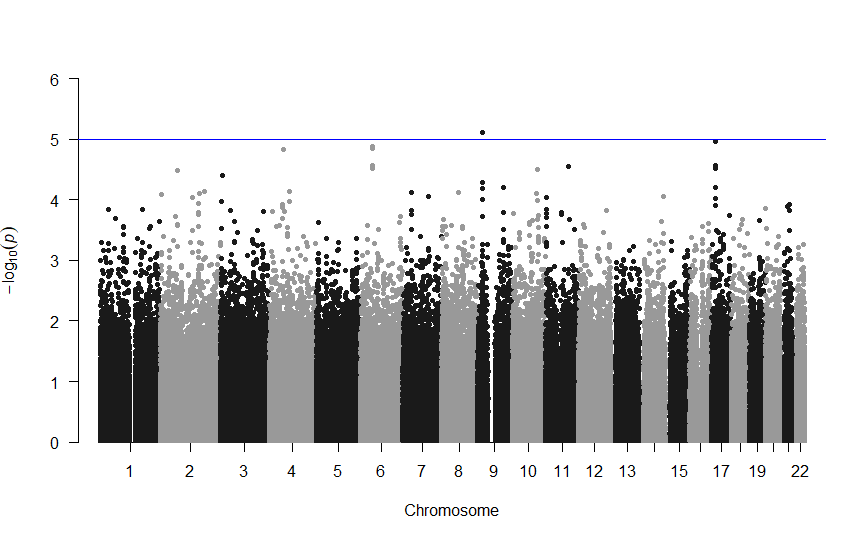
青色のラインはp値が1.0×10-5のレベルであり、suggestive level(有意ではないが、なんらかの関連がありそうなレベル)を示している。
ここにはないけど、赤色のラインはp値が5.0×10-8のレベルであり、GWASでな有意レベルを示している。
最終スクリプト
# Rをキレイにしておく
rm(list = ls())
# ライブラリの読み込み
library(qqman)
library(data.table)
# データの読み込みと内容の確認
DF <- fread("Yang_YoungOnsetHypertension_Illumina550_SBAS_rawpv.csv",
stringsAsFactors = F, header = T)
str(DF)
# マンハッタンプロットの描画
manhattan(DF, chr = "Chromosome", #染色体番号のカラムを指定する。
bp = "Physical_position", #染色体位置のカラムを指定する。
p = "CLR_C_BMI_pv", #p値のカラムを指定する。
snp = "dbSNP_RS_ID") #SNP名のカラム(rs番号)を指定する。