🌾本記事は下記の環境に基づいて執筆されました
- macOS Mojave (ver. 10.14.6)
-- R (ver. 3.6.2)
--- tidyverse (ver. 1.3.0); patchwork (ver. 1.0.0)
🌾本記事の概要
# バイオ系wet研究者が
# Rのtidyverseパッケージを利用して
# 実際の実験データを解析する
はじめに
本記事はRあるいはtidyverseパッケージについて詳細に解説することを目的としていません。それらについては,他の多くの記事や書籍がとても丁寧に解説してくれています。
特に自分は松村優哉 et al.『RユーザーのためのRStudio [実践] 入門』にかなり助けられました。
ただそれらの記事や書籍の多くは,irisやmpgのようなサンプルデータセットを利用して解説を行っており,バイオ系wet研究者 (特に蛋白質関係の分析が専門) な自分には少しとっつきにくかった (実用までの距離を感じた) ように思います。
そういった視点でネット上の解説記事を眺めてみると,実際の実験データを利用して解説している記事って案外少ないんじゃないかと思います。
やっぱり実験データはおいそれと公開できないからですかね。
(読者層も限定されちゃいますしね 笑)
そこで本記事 (とその続編) では,私が大学/大学院時代に測定したけどボツった実験データの中でも公開してなんら問題ないデータを活用して,研究の中で行っている実作業のログ (に近いもの) を共有できたら良いなと考えています。
「ああ,こういう使い方もできるのね」という感じで,誰かの (特に自分と専門が近い人たちの) モチベーションにつながることがあれば,それはとっても嬉しいなって。
Rおよびtidyverseを使い始めたきっかけ
最も有名なグラフ作成ソフトは何かと訊かれれば,それはもうみんな大好きMS Excel様になるわけですが,正直言って私はMS Excelでグラフを描くのが大の苦手です。特に発表資料に使うような「きれいな」図をMS Excelで作る自信はこれっぽっちもないですし,そんな作業をしたいとはこれっぽっちも思いません。 大学/大学院まではどうしていたかというと,Igor Pro (WaveMetrics) という有償のソフトウェアを利用していました。実験データの解析 (フィッティングなど) にもこいつは非常に活躍してくれたものです。 が,2019年の3月に無事学位を取得して (隙あらば……) 名残惜しくも大学を去り,企業に勤めることになった今,残念ながらIgor Proくんを使うことはできなくなりました。もちろん部署の予算で購入することはできましたが,ライセンスの関係上他の人に解析結果を共有することが難しいなと考え断念しました。 という経緯で,Igor Proの代わりになる「きれいな」グラフが描けて,実験の解析もできて,なおかつオープンな環境がないかなと考えていたとき,白羽の矢が立ったのが(余談ですので,興味のない方はすっ飛ばしてください)
Rとtidyverseだったわけです。今を時めくPythonちゃんにしなかった理由は (自主規制) です。
本記事の構成
本記事は
- 実験の背景説明
- 実験データの説明
- 実験データの解析
で構成されています。『実験の背景説明』は多くの人にとって不要かもしれませんが,ただ書きたかったのです (すみません)。
1. 蛋白質のUV–Visスペクトル
1.1. 背景を語る
蛋白質関係の分析で最もシンプルな測定といえば,UV–Visスペクトル測定ではないでしょうか。
特に蛋白質濃度を定量する際には,UV–Visスペクトル測定が広く用いられています。
フェニル基をもつフェニルアラニン,フェール基をもつチロシンやインドール基をもつトリプトファンも脂肪族側鎖と同様に疎水性で,蛋白質の内部に多く存在する。蛋白質を定量するときに280 nmの紫外吸収を用いることが多いが,この吸収はほとんどがトリプトファンとチロシンの吸収によるものである。
——有坂文雄『バイオサイエンスのための 蛋白質科学入門』より
他にもBCA法なんかで定量したりもできますが,BCA法はどうしても値がブレやすい (実験が下手とも言う) ので,自分はUV–Visスペクトルから濃度定量することが多いです。
UV–Visスペクトルから濃度を求める際には,いわゆるランベルト・ベールの法則を利用します。
$$A_{280} = \epsilon_{280} c l$$
ここで$A_{280}$,$\epsilon_{280}$,$c$,および$l$は,それぞれ波長280 nmにおける吸光度 (Abs.),波長280 nmにおけるモル吸光係数,モル濃度,および光路長 (通常は1 cm) を表しています。
蛋白質の$\epsilon_{280}$は,蛋白質のアミノ酸配列から容易に推定することが可能です。実験的に求めていたこともありましたが,結局BCA法を経ることになってイマイチなので,ここ数年はもっぱらアミノ酸配列から求めた理論値を使用しています。
ちなみに大学/大学院時代は,$\epsilon_{280}$の推定にProtParamをよく利用していました。
ラボによっては蛋白質の濃度定量の際に$A_{280}$だけしか測定しないところもあるようですが,400 nmより長波長側をみると凝集の度合いが確認できたり,大腸菌から持ち込まれた蛋白質以外の低分子 (例えばヘムなど) の吸収が観察されることもあります。なので,自分としては新しく扱う蛋白質の場合は,なるべく広範囲のUV–Visスペクトルを測定するようにしています。
1.2. 実験データについて
今回用いる実験データをGoogleスプレッドシートにアップしました。
https://docs.google.com/spreadsheets/d/1cki05r2u6X6dXOn1ziXMqtcKg1o6IwgV9dlD7zqQ0jw/edit?usp=sharing

以降はこのデータがprotein.xlsxのSheet1に記録されているものとして説明を行います。
.csvファイル等ではなくあえて.xlsxファイルを用いる理由ですが,結局他の実験者からデータを貰う場合はほとんどがMS Excelなんですよね。あと,最低限のデータ加工については,MS Excelで行ってしまう方が早いと考えています。自分が扱う実験データは,基本的にMS Excelで編集できる程度のサイズですからね……。
本データの内容は,PBS溶液 (pH 7.4) に溶解した濃度未知な某蛋白質のUV–Visスペクトルです。本蛋白質の$\epsilon_{280}$については,アミノ酸配列から25,900 M−1 cm−1と推定されています。
本UV–Visスペクトルは,UV–Vis分光光度計UV-2600 (Shimadzu) を用いて測定されました。サンプルセルには光路長1 cmの石英セルを用いました。
一般に光路長1 cmの分光光度計において信頼できるAbs.の範囲は,おおよそ0.2–2.0だと思われます (もちろん機種によりますが)。今回測定した蛋白質溶液は非常に濃いと予想されたため,原液を20倍希釈してスペクトルを測定しました。
1.3. 実験データの解析
それでは実験データの解析に移ります。
はじめに,必要なパッケージのインストールと読み込みを行います。
# パッケージのインストール
install.packages("tidyverse")
# パッケージの読み込み
library(tidyverse)
library(readxl)
tidyverseには,今後使用する様々なパッケージ (例えばdplyrやggplot2など) が含まれています。
readxlはExcelファイルを読み込むために使用します。
readxlはtidyverseに含まれているため個別でのインストールは不要ですが,使用するためには個別に読み込む必要があります。
次に,Excelファイルにまとめた測定データ (protein.xlsx) をインポートしてprot_rawに格納します。
# 測定データをExcelファイルからインポート
prot_raw <- read_excel("protein.xlsx", sheet = "Sheet1")
prot_rawの中身はこんな感じです。いい感じに読み込めていますね。
> prot_raw
# A tibble: 521 x 2
wavelength_nm abs_protein_x20
<dbl> <dbl>
1 240 1.52
2 240. 1.39
3 241 1.28
4 242. 1.17
5 242 1.07
6 242. 0.979
7 243 0.898
8 244. 0.829
9 244 0.769
10 244. 0.714
# … with 511 more rows
ここで,測定の際に蛋白質溶液を20倍希釈したことを考慮し,蛋白質Abs.の実測値であるabs_protein_x20列を20倍したabs_protein列を新たに作成します。
これにはdplyrパッケージのmutate()関数を使用します。
# 蛋白質のAbs.実測値を20倍した列を追加
prot <- mutate(prot_raw, abs_protein = abs_protein_x20 * 20)
生データに対してデータ処理を行ったので,prot_rawあらためprotというデータフレームに格納しています。
上記の処理は%>% (パイプ演算子) を利用すると次のように書けます (慣れましょう)。
# 蛋白質のAbs.実測値を20倍した列を追加
prot <- prot_raw %>%
mutate(abs_protein = abs_protein_x20 * 20)
protの中身はこんな感じです。いい感じにデータ編集できました。
> prot
# A tibble: 521 x 3
wavelength_nm abs_protein_x20 abs_protein
<dbl> <dbl> <dbl>
1 240 1.52 30.5
2 240. 1.39 27.8
3 241 1.28 25.5
4 242. 1.17 23.3
5 242 1.07 21.4
6 242. 0.979 19.6
7 243 0.898 18.0
8 244. 0.829 16.6
9 244 0.769 15.4
10 244. 0.714 14.3
# … with 511 more rows
それではいよいよ蛋白質のUV–Visスペクトルをプロットしてみましょう。
グラフの作成にはggplot2パッケージを使用します。
シンプルに書くと次のようなコードになります。
# 蛋白質のUV-Visスペクトルをプロット
prot %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_protein)) +
geom_line()
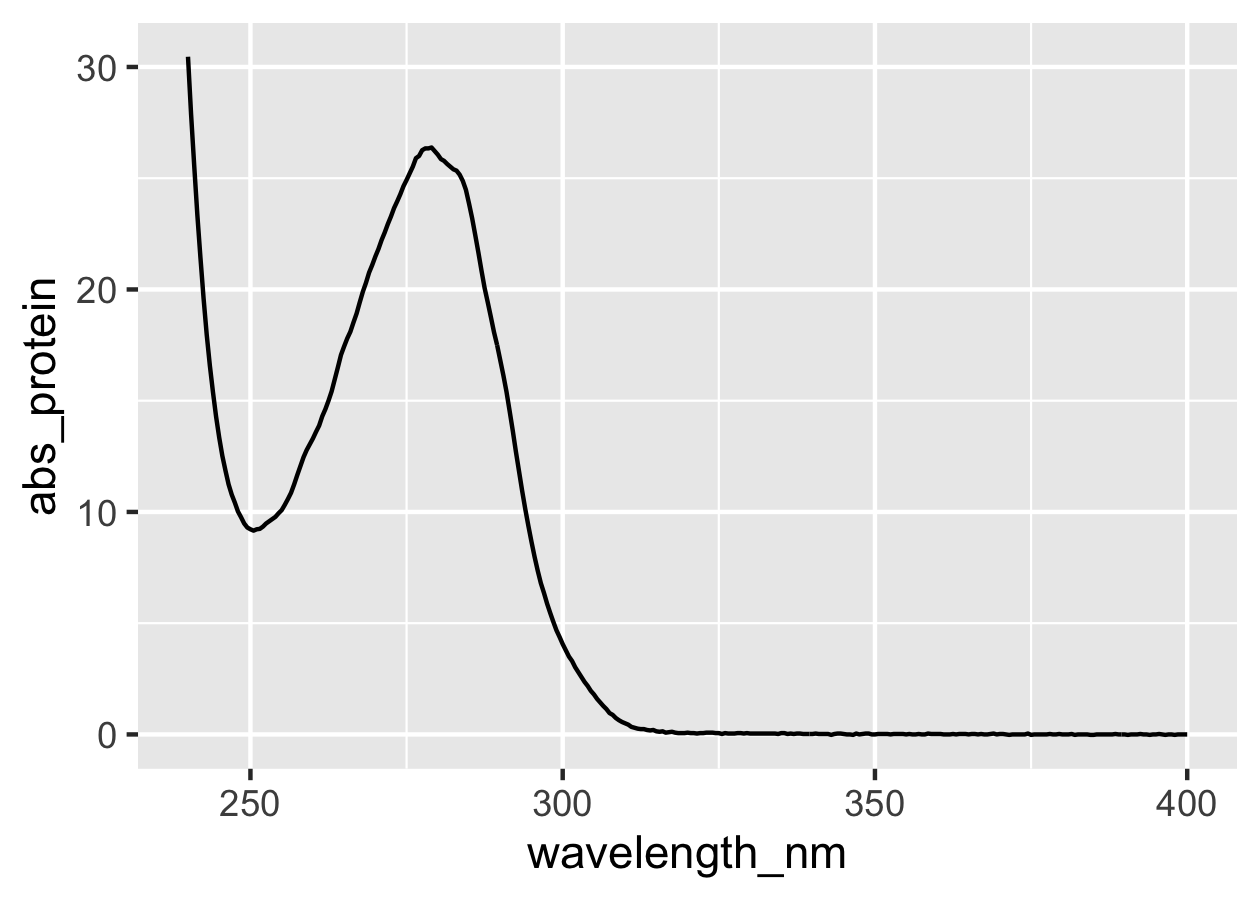
はい,プロットできました。
ここから少し描画の設定をいじってみます。
geom_line()においてcolourおよびsizeを指定することで,プロットの色および線の太さをそれぞれ変更します。
scale_x_continuous()を指定することでx軸の取り方を変更します。
(蛋白質のUV–Visスペクトルでは280 nmの値が重要)
labs()で軸名を変更します
# 蛋白質のUV-Visスペクトルをプロット
prot %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_protein)) +
geom_line(colour = "#00ACC1", size = 1.2) +
scale_x_continuous(breaks = seq(240, 400, 40), limits = c(240, 400)) +
labs(x = "Wavelength (nm)", y = "Absorbance")
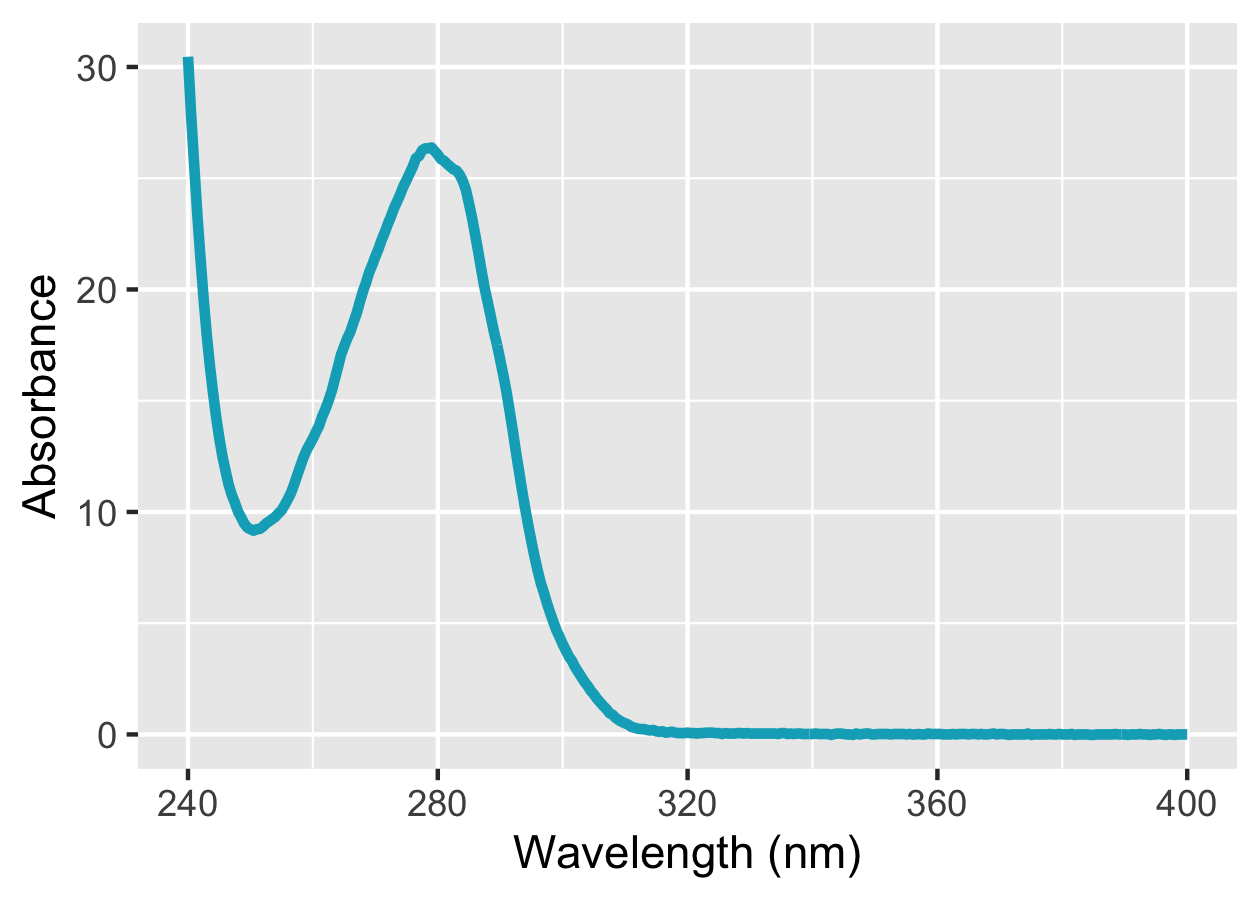
だいぶ良い感じになりました。ただ,ggplot2のデフォルトテーマ (グレー背景) だと少しモダン過ぎるように感じるので,theme_set()関数を用いて自分なりにテーマを変更します。
# グラフのテーマ設定 (macOSの場合)
theme_set(
theme_minimal(base_size = 20, base_family = "Helvetica Neue Bold") +
theme(
panel.grid.major = element_line(colour = "#BDBDBD"),
panel.grid.minor = element_blank(),
axis.title.x = element_text(margin = margin(7, 0, 0, 0)),
axis.title.y = element_text(margin = margin(0, 8, 0, 0)),
axis.text = element_text(colour = "#424242"),
plot.background = element_rect(fill = "transparent", color = NA),
legend.title = element_blank(),
legend.justification = "top"
)
)
# 蛋白質のUV-Visスペクトルをプロット
prot %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_protein)) +
geom_line(colour = "#00ACC1", size = 1.3) +
scale_x_continuous(breaks = seq(240, 400, 40), limits = c(240, 400)) +
labs(x = "Wavelength (nm)", y = "Absorbance")
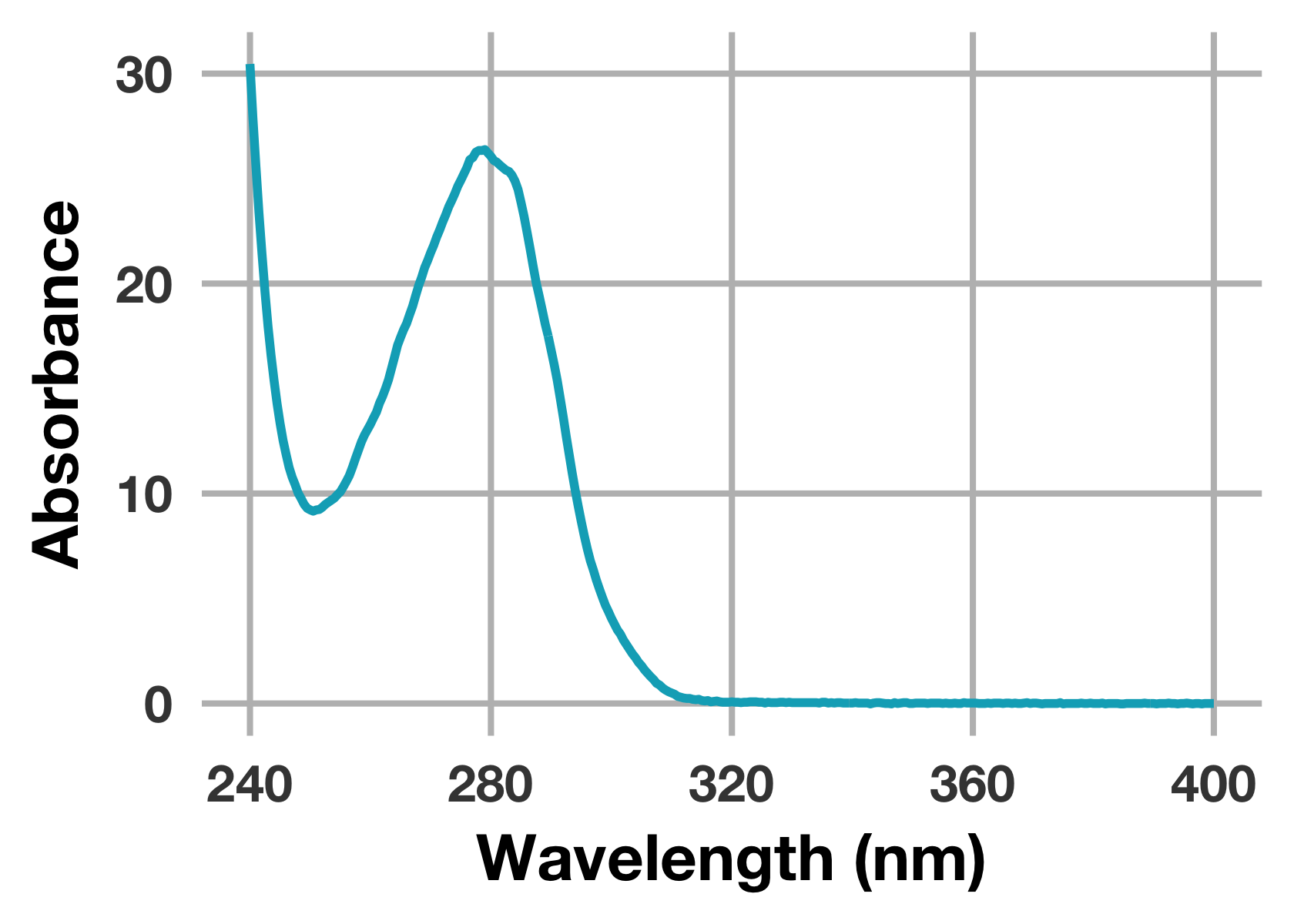
はい,良い感じにグラフが描画できました。もっとクラシカルな表現も可能ですが,自分はいろいろ試行錯誤した結果この設定に落ち着きました (少しモダンな方が社内で目立つという浅はかな考え……)。
themeの設定については細かく説明しません。複雑なことはしていないので,興味のある方は一行ずつ調べてみてください。
ちなみにWindowsの場合はフォント指定の部分のみ少し変わります。
# グラフのテーマ設定 (Windowsの場合)
windowsFonts(SG = windowsFont("Segoe UI Bold")) # この行が違う
theme_set(
theme_minimal(base_size = 20, base_family = "SG") + # この行も少し違う
theme(
panel.grid.major = element_line(colour = "#BDBDBD"),
panel.grid.minor = element_blank(),
axis.title.x = element_text(margin = margin(7, 0, 0, 0)),
axis.title.y = element_text(margin = margin(0, 8, 0, 0)),
axis.text = element_text(colour = "#424242"),
plot.background = element_rect(fill = "transparent", color = NA),
legend.title = element_blank(),
legend.justification = "top"
)
)
グラフが書けたのでggsave()関数を用いて画像として保存しましょう。
# グラフを保存
ggsave("protein.png",
dpi = 300, width = 5.6, height = 4,
bg = "transparent")
ちなみに,本蛋白質の$\epsilon_{280}$は25,900 M−1 cm−1であることを思い出すと,ランベルト・ベールの法則から次のようにして蛋白質濃度を求めることができます。
# 蛋白質濃度を計算
prot %>%
filter(wavelength_nm == 280) %>%
mutate(conc_mM = abs_protein / 25900 * 1000)
# 出力結果
# A tibble: 1 x 4
wavelength_nm abs_protein_x20 abs_protein conc_mM
<dbl> <dbl> <dbl> <dbl>
1 280 1.30 26.1 1.01
蛋白質濃度が1.0 mMと求まりましたね。
それでは続いて,低分子化合物のUV–Visスペクトル測定データの解析に移ります。
2. 化合物のUV–Visスペクトル (モル吸光係数の決定)
2.1. 背景を語る
低分子化合物は蛋白質と異なり,化合物ごとに様々なUV–Visスペクトルを示します。したがって,化合物の濃度定量を分光学的に行うためには,化合物ごとに固有の$\epsilon$を実験的に決定する必要があります。
実験的に$\epsilon$を決定するといっても,それほど難しい作業ではありません。
まず,化合物を精密天秤で秤量し,有機溶媒 (基本的にはDMSO) に溶解させます。次に,秤量値と化合物の分子量からこのストック溶液の濃度を決定し,あとは段階希釈したサンプルについてそれぞれUV–Visスペクトルを測定します。得られたUV–VisスペクトルのピークトップにおけるAbs.を希釈率から計算した化合物の理論濃度に対してプロットし,線形回帰によって傾きを求めればピークトップの波長における$\epsilon$が求まります。
出身ラボでは新規の化合物を購入した際は,はじめに100% DMSO中における$\epsilon$を決定する習慣がありました。$\epsilon$を一度決定してしまえば,たとえ吸湿や量的問題から精度に欠ける秤量になったとしても,UV–Visスペクトルを測定すれば濃度定量できるという安心感があるからです。
ただし,溶媒によって$\epsilon$が大きく変わることはざらですし,$\epsilon$決定時の秤量が不正確だと以降の実験がすべてデタラメになってしまいます。あとは購入ロットで少し変わったり……。
2.2. 実験データについて
今回用いる実験データをGoogleスプレッドシートにアップしました。
https://docs.google.com/spreadsheets/d/1QDpoPPa0p7IX9-hrqCUXobhNY8QvXVTyfJmyV87FXEo/edit?usp=sharing
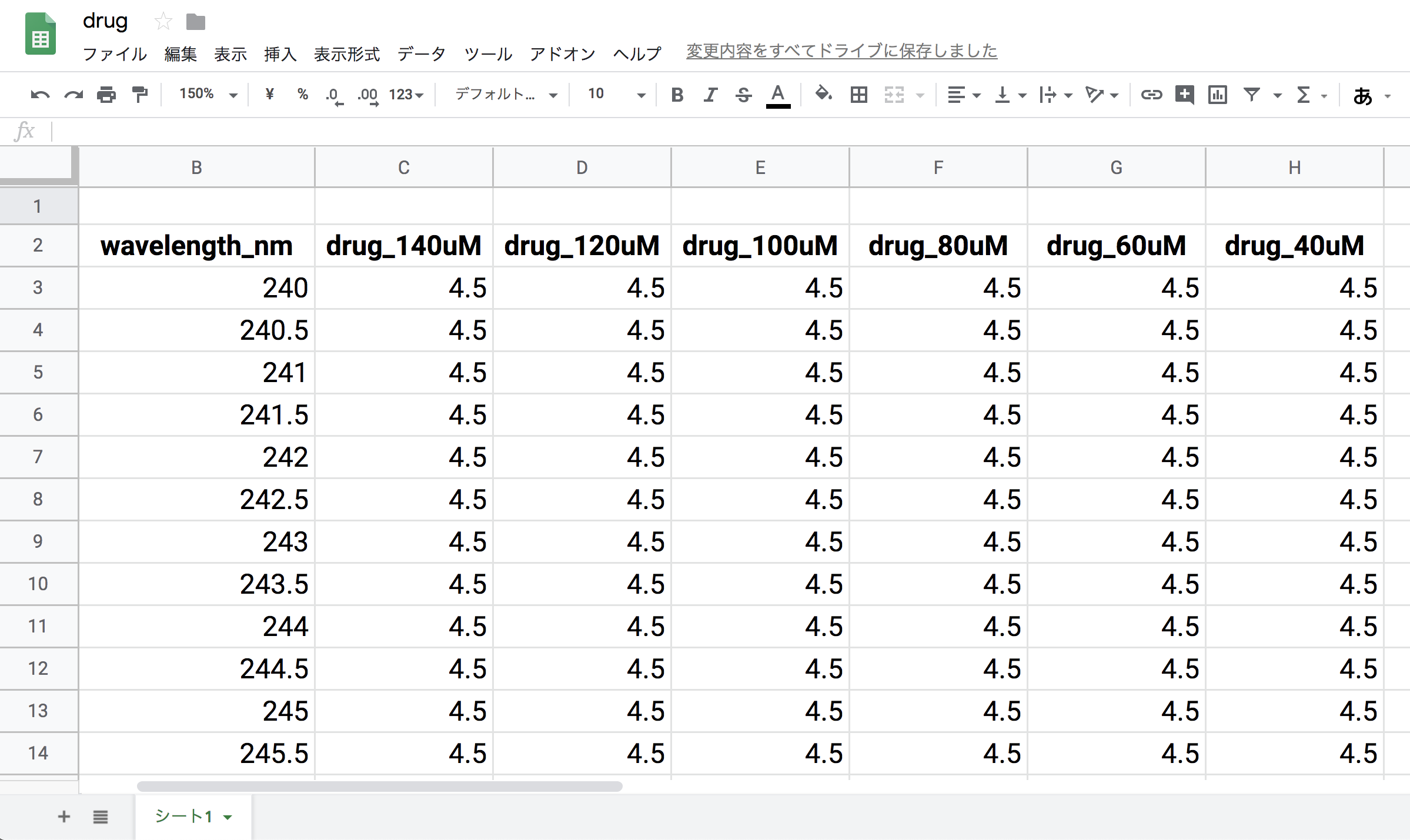
以降はこのデータがdrug.xlsxのSheet1に記録されているものとして説明を行います。
本データの内容は,DMSOに溶解した濃度既知 (40–140 μM) な某薬剤化合物のUV–Visスペクトルです。
本UV–Visスペクトルは,UV–Vis分光光度計DU 800 (Beckman Coulter) を用いて測定されました。サンプルセルには光路長1 cmの石英セルを用いました。
2.3. 実験データの解析
さきほどと同様に,Excelファイルにまとめた測定データ (drug.xlsx) をインポートしてprot_rawに格納します。
# 実験データをExcelファイルからインポート
drug_raw <- read_excel("drug.xlsx", sheet = "Sheet1")
drug_rawの中身はこんな感じです。いい感じに読み込めていますね。
> drug_raw
# A tibble: 321 x 7
wavelength_nm drug_140uM drug_120uM drug_100uM drug_80uM drug_60uM drug_40uM
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 240 4.5 4.5 4.5 4.5 4.5 4.5
2 240. 4.5 4.5 4.5 4.5 4.5 4.5
3 241 4.5 4.5 4.5 4.5 4.5 4.5
4 242. 4.5 4.5 4.5 4.5 4.5 4.5
5 242 4.5 4.5 4.5 4.5 4.5 4.5
6 242. 4.5 4.5 4.5 4.5 4.5 4.5
7 243 4.5 4.5 4.5 4.5 4.5 4.5
8 244. 4.5 4.5 4.5 4.5 4.5 4.5
9 244 4.5 4.5 4.5 4.5 4.5 4.5
10 244. 4.5 4.5 4.5 4.5 4.5 4.5
# … with 311 more rows
次のようにこのまま強引にプロットすることも可能です。
drug_raw %>%
ggplot(mapping = aes(x = wavelength_nm)) +
geom_line(mapping = aes(y = drug_40uM), colour = "#8D6E63", size = 1.3) +
geom_line(mapping = aes(y = drug_60uM), colour = "#AB47BC", size = 1.3) +
geom_line(mapping = aes(y = drug_80uM), colour = "#00ACC1", size = 1.3) +
geom_line(mapping = aes(y = drug_100uM), colour = "#43A047", size = 1.3) +
geom_line(mapping = aes(y = drug_120uM), colour = "#FB8C00", size = 1.3) +
geom_line(mapping = aes(y = drug_140uM), colour = "#EF5350", size = 1.3) +
labs(x = "Wavelength (nm)", y = "Absorbance")
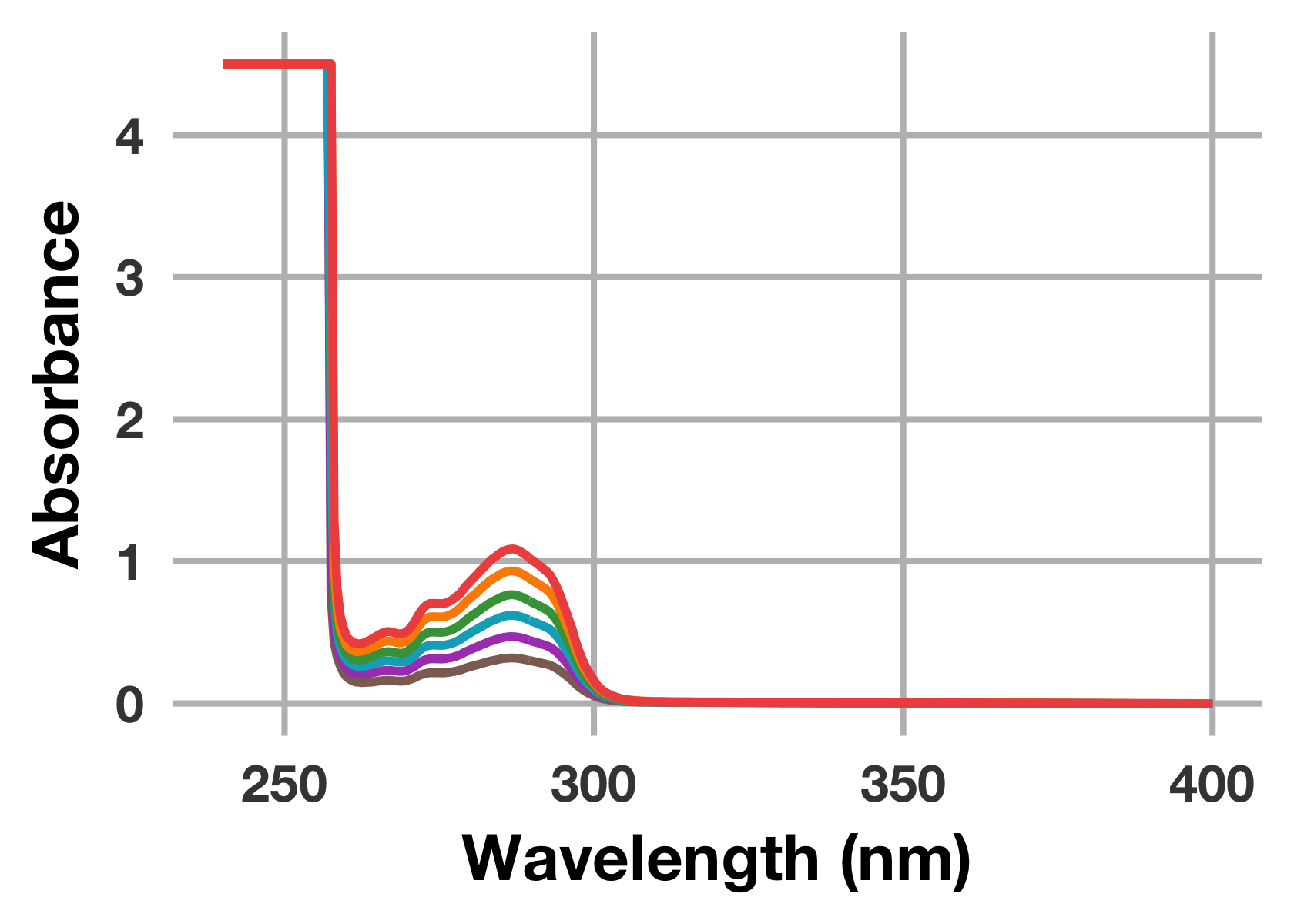
場合によってはこのように強引にプロットする方が楽かもしれませんが,ちょっとスマートではありませんし,このままだと自動でlabel (凡例) が表示されません。そこで,プロットを行う前に読み込んだデータをtidy dataに変換する作業を行います。
tidy dataというと少し難しく感じられますが,今回の場合だと横長データを縦長データに変換するだけです (下図参照)。変換にはdplyrパッケージのpivot_longer()関数を利用します。

# 実験データをtidy dataに変換
drug_long <- drug_raw %>%
pivot_longer(col = -wavelength_nm, names_to = "conc_drug_uM", values_to = "abs_drug")
drug_longの中身はこんな感じです。うまく縦長データに変換できました。
> drug_long
# A tibble: 1,926 x 3
wavelength_nm conc_drug_uM abs_drug
<dbl> <chr> <dbl>
1 240 drug_140uM 4.5
2 240 drug_120uM 4.5
3 240 drug_100uM 4.5
4 240 drug_80uM 4.5
5 240 drug_60uM 4.5
6 240 drug_40uM 4.5
7 240. drug_140uM 4.5
8 240. drug_120uM 4.5
9 240. drug_100uM 4.5
10 240. drug_80uM 4.5
# … with 1,916 more rows
pivot_longer()については
『【tidyr】gather?, spread? もう古い。時代はpivot』
を参考にさせていただきました。
次にdplyrパッケージのmutate()関数を用いてconc_drug_uM列を濃度の値に変換します。
さらにarrange()関数によってwavelength_nmおよびconc_drug_uMを昇順でソートします。
# conc_drug_uM列を対応する濃度の値に変換
drug_long <- drug_long %>%
mutate(conc_drug_uM = case_when(
str_detect(conc_drug_uM, fixed("_40")) ~ 40,
str_detect(conc_drug_uM, fixed("_60")) ~ 60,
str_detect(conc_drug_uM, fixed("_80")) ~ 80,
str_detect(conc_drug_uM, fixed("_100")) ~ 100,
str_detect(conc_drug_uM, fixed("_120")) ~ 120,
str_detect(conc_drug_uM, fixed("_140")) ~ 140)) %>%
arrange(wavelength_nm, conc_drug_uM)
これはなんというか,もっとスマートな書き方がありそうですね。
drug_longの中身はこのように変換されました。
> drug_long
# A tibble: 1,926 x 3
wavelength_nm conc_drug_uM abs_drug
<dbl> <dbl> <dbl>
1 240 40 4.5
2 240 60 4.5
3 240 80 4.5
4 240 100 4.5
5 240 120 4.5
6 240 140 4.5
7 240. 40 4.5
8 240. 60 4.5
9 240. 80 4.5
10 240. 100 4.5
# … with 1,916 more rows
それではtidy dataに変換したデータを用いて,化合物のUV–Visスペクトルをプロットしてみます。
# 化合物のUV-Visスペクトルをプロット
drug_long %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_drug, colour = factor(conc_drug_uM))) +
geom_line(size = 1.3) +
labs(x = "Wavelength (nm)", y = "Absorbance", colour = "Conc. (μM)") +
theme(legend.title = element_text(size = 16))
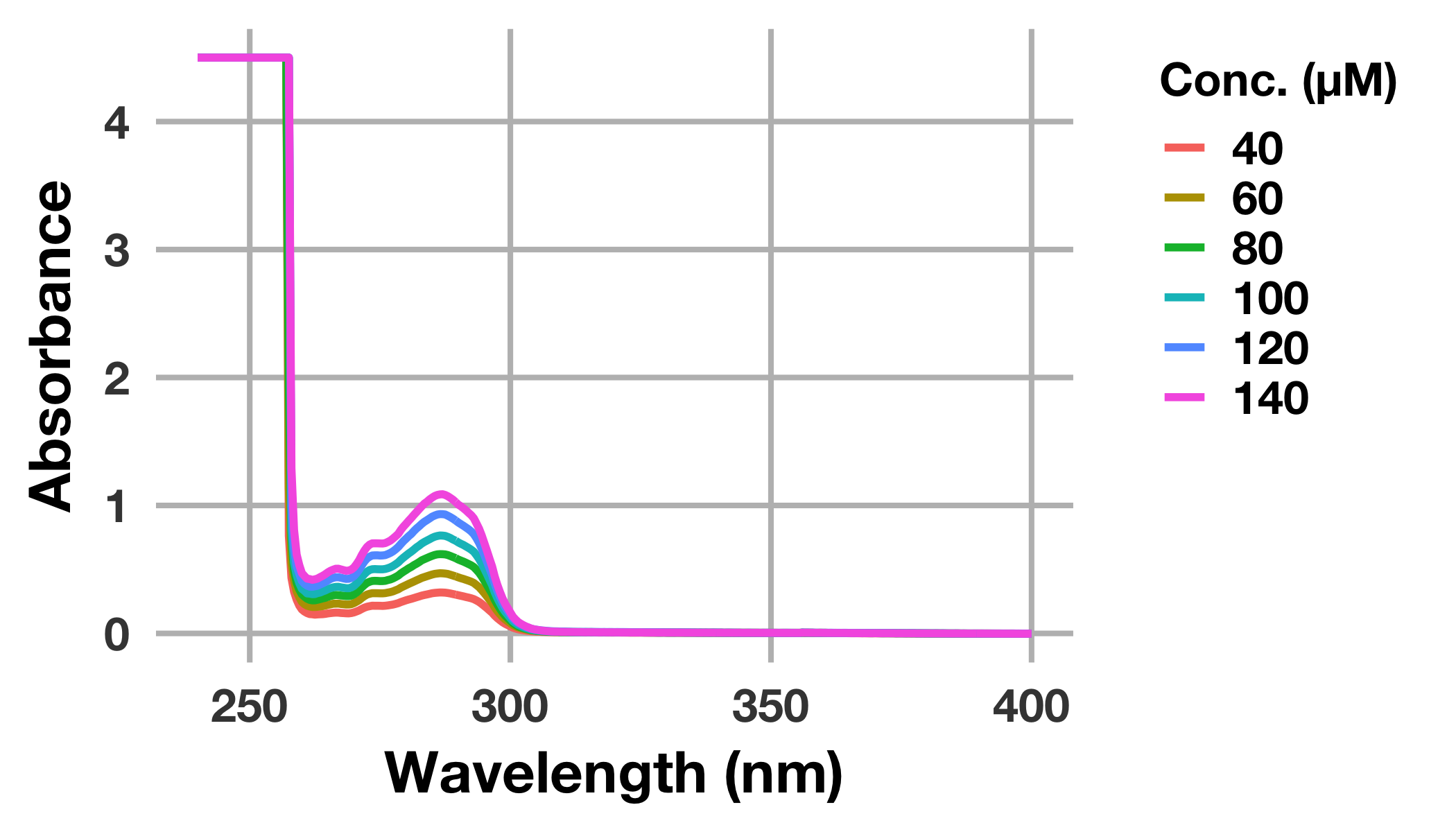
labelの表示や配色が自動で行われました。
ggplot(mapping = aes())においてcolourにfactor(conc_drug_uM)を渡していますが,これはconc_drug_uMを離散変数と認識させるためです。
グラフからも判るように短波長側は溶媒であるDMSOの影響を強く受けるため,あらためて波長260–380 nmを描画してみましょう。
同時にscale_colour_manualを設定し,マニュアルでの配色を行ってみます。
# 化合物のUV-Visスペクトルをプロット
drug_long %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_drug, colour = factor(conc_drug_uM))) +
geom_line(size = 1.3) +
scale_colour_manual(values = c("#8D6E63", "#AB47BC", "#00ACC1",
"#43A047", "#FB8C00", "#EF5350")) +
scale_x_continuous(breaks = seq(260, 380, 20), limits = c(260, 380)) +
scale_y_continuous(breaks = seq(0, 1.2, 0.2), limits = c(0, 1.2)) +
labs(x = "Wavelength (nm)", y = "Absorbance", colour = "Conc. (μM)") +
theme(legend.title = element_text(size = 16))
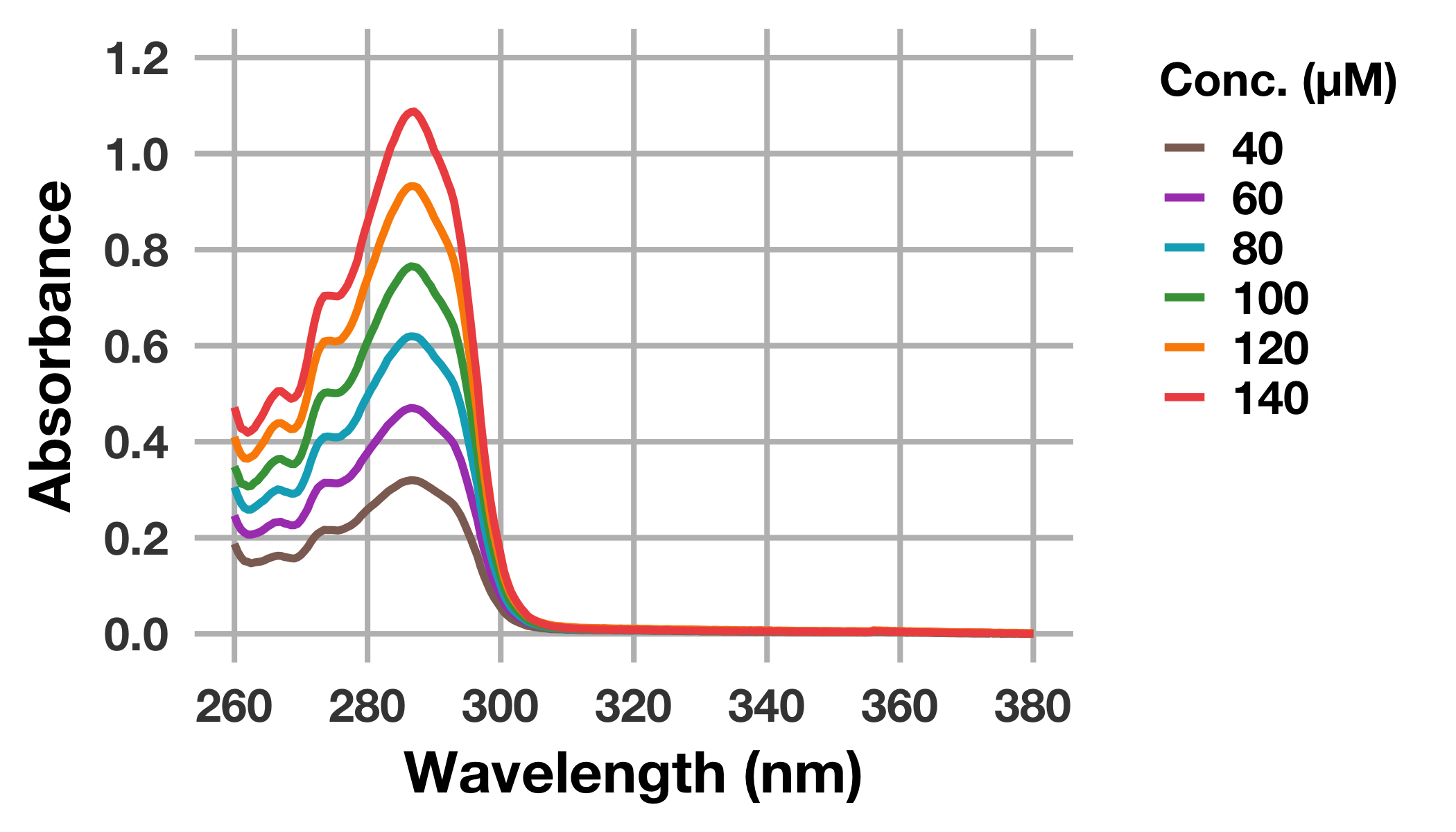
これで満足のいくUV–Visスペクトルが得られました。
それでは続いて,本薬剤化合物の$\epsilon$を求めてみましょう。
まず,各濃度のUV–VisスペクトルにおけるピークトップのAbs.を抽出します。
このとき,DMSOの影響を受ける短波長側 (本データでは240–260 nm) を除いた範囲でピークトップを抽出することに留意してください。
# 波長260-400 nmにおけるピークトップを抽出
peak_top <- drug_long %>%
filter(wavelength_nm >= 260) %>%
group_by(conc_drug_uM) %>%
slice(which.max(abs_drug))
slice(which.max())については
『本日の懺悔:dplyrでsliceをもっと活用しようね』
を参考にさせていただきました。
peak_topの中身を確認すると……
> peak_top
# A tibble: 6 x 3
# Groups: conc_drug_uM [6]
wavelength_nm conc_drug_uM abs_drug
<dbl> <dbl> <dbl>
1 286. 40 0.32
2 286. 60 0.470
3 286. 80 0.620
4 286. 100 0.765
5 286. 120 0.932
6 287 140 1.09
各濃度におけるピークトップのAbs.を抽出することができました。
ピークトップは286–287 nm付近のようです。さきほどのグラフを観かえすと,どうやら問題なさそうだなということが判ります。
続いて本データに対して線形回帰を実行し,$\epsilon_{286}$を決定します。
線形回帰にはlm()関数を用いて次のように行います。
# 線形回帰によるモル吸光係数 (e286) の決定
fit_e286 <- peak_top %>%
lm(abs_drug ~ 0 + conc_drug_uM, data = .)
線形回帰の結果をsummary()関数で確認します。
> summary(fit_e286)
Call:
lm(formula = abs_drug ~ 0 + conc_drug_uM, data = .)
Residuals:
1 2 3 4 5 6
0.009727 0.004990 -0.001047 -0.010383 0.001580 0.001743
Coefficients:
Estimate Std. Error t value Pr(>|t|)
conc_drug_uM 7.757e-03 2.901e-05 267.4 1.39e-11 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 0.00684 on 5 degrees of freedom
Multiple R-squared: 0.9999, Adjusted R-squared: 0.9999
F-statistic: 7.15e+04 on 1 and 5 DF, p-value: 1.388e-11
以上より,本化合物の$\epsilon_{286}$は7760 M−1 cm−1 (R2 = 0.9999) と求まりました (有効数字3桁)。
続いて線形回帰の結果を可視化してみましょう。
まずggplot2に読み込ませるために,線形回帰の結果を基に関数を定義します。
# 決定したe286を基に関数を作成
fit_curve_e286 <- function(x) {
e286 <- coef(fit_e286)[1]
return(e286 * x)
}
上記コードのfit_e286[1]は,線形回帰によって決定した$\epsilon_{286}$のことです。
それでは,ggplot2で可視化してみましょう。
# モル吸光係数に関する解析結果をプロット
peak_top %>%
ggplot(mapping = aes(x = conc_drug_uM, y = abs_drug)) +
stat_function(fun = fit_curve_e286, size = 1.3, colour = "black") +
geom_point(
shape = 21, color = "black", fill = "#00ACC1",
size = 5, stroke = 1.5) +
scale_x_continuous(breaks = seq(0, 180, 40), limits = c(0, 180)) +
scale_y_continuous(breaks = seq(0, 1.5, 0.4), limits = c(0, 1.5)) +
labs(x = "Conc. of drug (μM)", y = "Abs. at 286 nm")
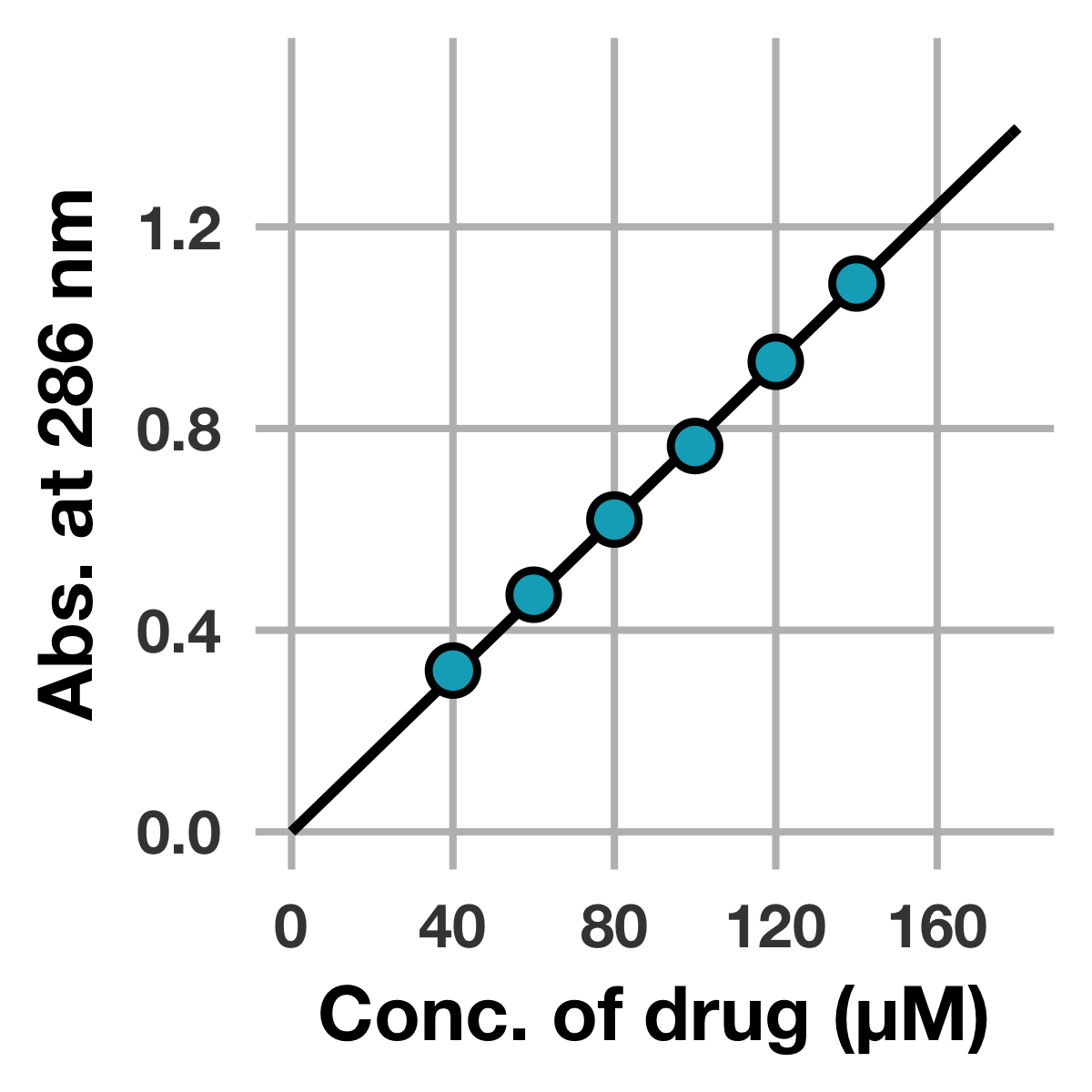
はい,できました!
グラフの中にテキストで$\epsilon_{286}$の値を追記することも可能ですが,正直Rで位置を調整するのは骨が折れるので,自分はMS PowerPoint上で済ませることが多いです (邪悪ですみません)。
最後に化合物のUV–Visスペクトルと線形回帰のグラフを並べて表示させてみましょう。
追加のパッケージpatchworkをインストール & ロードします。
# patchworkをインストール & ロード
install.packages("patchwork")
library(patchwork)
さきほど作成したUV–Visスペクトルと線形回帰のグラフを,それぞれg_drug_speおよびg_drug_e286というオブジェクトに格納し,patchworkを使って足し合わせます。
# 化合物のUV-Visスペクトルをプロット
g_drug_spe <- drug_long %>%
ggplot(mapping = aes(x = wavelength_nm, y = abs_drug, colour = factor(conc_drug_uM))) +
geom_line(size = 1.3) +
geom_vline(xintercept = 286.5, linetype = "dashed", size = 1) +
scale_colour_manual(values = c("#8D6E63", "#AB47BC", "#00ACC1",
"#43A047", "#FB8C00", "#EF5350")) +
scale_x_continuous(breaks = seq(260, 380, 20), limits = c(260, 380)) +
scale_y_continuous(breaks = seq(0, 1.2, 0.2), limits = c(0, 1.2)) +
labs(x = "Wavelength (nm)", y = "Absorbance", colour = "Conc. (μM)") +
theme(legend.title = element_text(size = 16))
# モル吸光係数に関する解析結果をプロット
g_drug_e286 <- peak_top %>%
ggplot(mapping = aes(x = conc_drug_uM, y = abs_drug)) +
stat_function(fun = fit_curve_e286, size = 1.3, colour = "black") +
geom_point(
shape = 21, color = "black", fill = "#00ACC1",
size = 5, stroke = 1.5) +
scale_x_continuous(breaks = seq(0, 180, 40), limits = c(0, 180)) +
scale_y_continuous(breaks = seq(0, 1.5, 0.4), limits = c(0, 1.5)) +
labs(x = "Conc. of drug (μM)", y = "Abs. at 286 nm")
# patchworkパッケージを用いてグラフをがっちゃんこ
g_drug_spe + g_drug_e286 +
plot_annotation(tag_levels = "A")

おお! これはなんというかすごい (記事を書くまでpatchworkを使ったことがなかった人)。
論文で使用するFigureを作成するのに非常に便利なのではないだろうか。積極的に使っていこう……。
これはおまけ程度ですが,g_drug_speについてはgeom_vline()を追記し,ピークトップの波長を示す破線を追加しています。
patchworkについては
『patchworkを使って複数のggplotを組み合わせる』
を参考にさせていただきました。
以上で今回はおしまいです。
次回予告
次回はトリプトファン蛍光消光試験による蛋白質–低分子の相互作用解析を予定しています。