*コマンドにミスが見つかりましたので直しましたが、内容やバージョンは変えておりません。更新はされていますが、内容は2020年1月に書いたものです。最新情報ではありませんのであしからず。
StringTieからのBallgownは固定じゃない
Nature Protocolがあまりにも分かりやすいので、僕は「StringTieとBallgownはセット!StringTieを使ったら、Ballgownでしょ。」などど考えていたが、マニュアルにも記載があるように、DESeq2やedgeRというソフトも計算に利用できるらしい。今回はStringTieで出力されたGTFファイルからDES
eq2用のファイルを書き出し、最終的にDESeq2のマニュアルを参考に、データ解析をする方法をやってみたいと思う。DESeq2のマニュアルをざっと見たがヒートマップが一番おしゃれな感じがするので、今回はヒートマップを書くことに決めた。なお、使用するデータは先輩が練習で動かしたデータ。何の生物でやられたデータなのかすら知らない。
参考: http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual
StringTieのGTFファイルを書き換える
まず、これをするにはprepDE.pyが必要となる。ただし、prepDE.pyはPython2で書かれているので、Python2を動かせるようにする。当方はPython2を入れてないので、Homebrewに入れた。ワーキングディレクトリ内にprepDE.pyを入れておくことをお忘れなく。
$ brew install python2
Python2を利用する際は以下コマンドで利用できる。
python2 hoge.py
その後、sample_list.txtを作成する (作戦1に例を記載)。なお、このテキストファイルは必須アイテムではない。こんなテキストファイル作るのめんどくさいぜって人は作戦2を利用する。
作戦1
txtファイルの中身
ERR188021 <PATH_TO_ERR188021.gtf>
ERR188023 <PATH_TO_ERR188023.gtf>
ERR188024 <PATH_TO_ERR188024.gtf>
ERR188025 <PATH_TO_ERR188025.gtf>
ERR188027 <PATH_TO_ERR188027.gtf>
ERR188028 <PATH_TO_ERR188028.gtf>
ERR188030 <PATH_TO_ERR188030.gtf>
ERR188033 <PATH_TO_ERR188033.gtf>
ERR188034 <PATH_TO_ERR188034.gtf>
ERR188037 <PATH_TO_ERR188037.gtf>
使用するコマンド
python2 prepDE.py -i sample_list.txt
作戦2
作戦2は要はデフォルトで決められてるパス内にファイルを配置するということである。ワーキングディレクトリ内にballgownディレクトリを作成し、その中にさらに各サンプルごとのディレクトリを作る。StringTieでデータ作るとこんな感じになってるはずなので、作戦2の方が早そうである。他のデータが混じってても、普通に動いた。
# GTFファイルの配置方法
./ballgown/sample1/sample1.gtf
./ballgown/sample2/sample2.gtf
./ballgown/sample3/sample3.gtf
使用するコマンド
python2 prepDE.py
すると、2個のファイルができる。
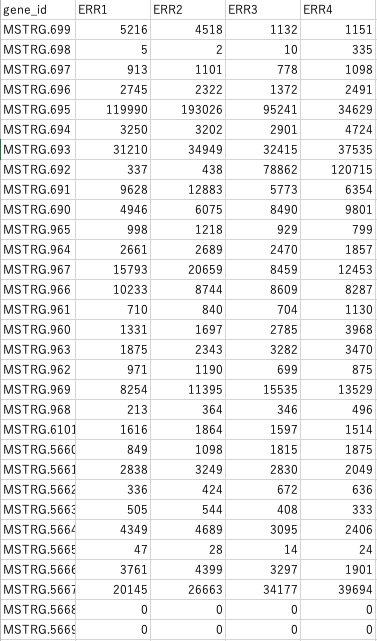
それぞれgene_count_matrix.csv、transcript_count_matrix.csvという名前で、その名の通り遺伝子単位のリードカウントと転写産物のリードカウントがTableになっている。
データ解析
先のStringTieのサイトにやり方が載っているので、このままやりたいと思う。
stall R and DESeq2. Upon installing R, install DESeq2 (Rを起動、DESeq2のインストール)
> source("https://bioconductor.org/biocLite.R")
> biocLite("DESeq2")
Import DESeq2 library in R (ライブラリの起動)
> library("DESeq2")
Load gene(/transcript) count matrix and labels
> countData <- as.matrix(read.csv("gene_count_matrix.csv", row.names="gene_id"))
> colData <- read.csv(PHENO_DATA, row.names=1)
今回、Phenodata fileとして先輩から預かったcsvを利用した。
# phenodata.csvの中身
ids,oxidate
ERR1,Positive
ERR2,Positive
ERR3,Negative
ERR4,Negative
Check all sample IDs in colData are also in CountData and match their orders (phenodata.csvとサンプルが噛み合うか確認)
> all(rownames(colData) %in% colnames(countData))
[1] TRUE
> countData <- countData[, rownames(colData)]
> all(rownames(colData) == colnames(countData))
[1] TRUE
Create a DESeqDataSet from count matrix and labels (DESeqDataSetを作成)
> dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData, design = ~ CHOOSE_FEATURE)
注意ポイント design = ~ CHOOSE_FEATUREの~は必要。なので、例えば今回ならdesign = ~ oxidateとする。
> dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData, design = ~ oxidate)
Run the default analysis for DESeq2 and generate results table (デフォルトでDESeq2による解析を行い、結果を表にまとめる)
> dds <- DESeq(dds)
> res <- results(dds)
Sort by adjusted p-value and display (P-Valueによって結果をソートする)
> (resOrdered <- res[order(res$padj), ])
Dispension Plotをしてもいい。
> png("qc-dispersions.png", 1000, 1000, pointsize=20)
> plotDispEsts(dds, main="Dispersion plot")
> dev.off()
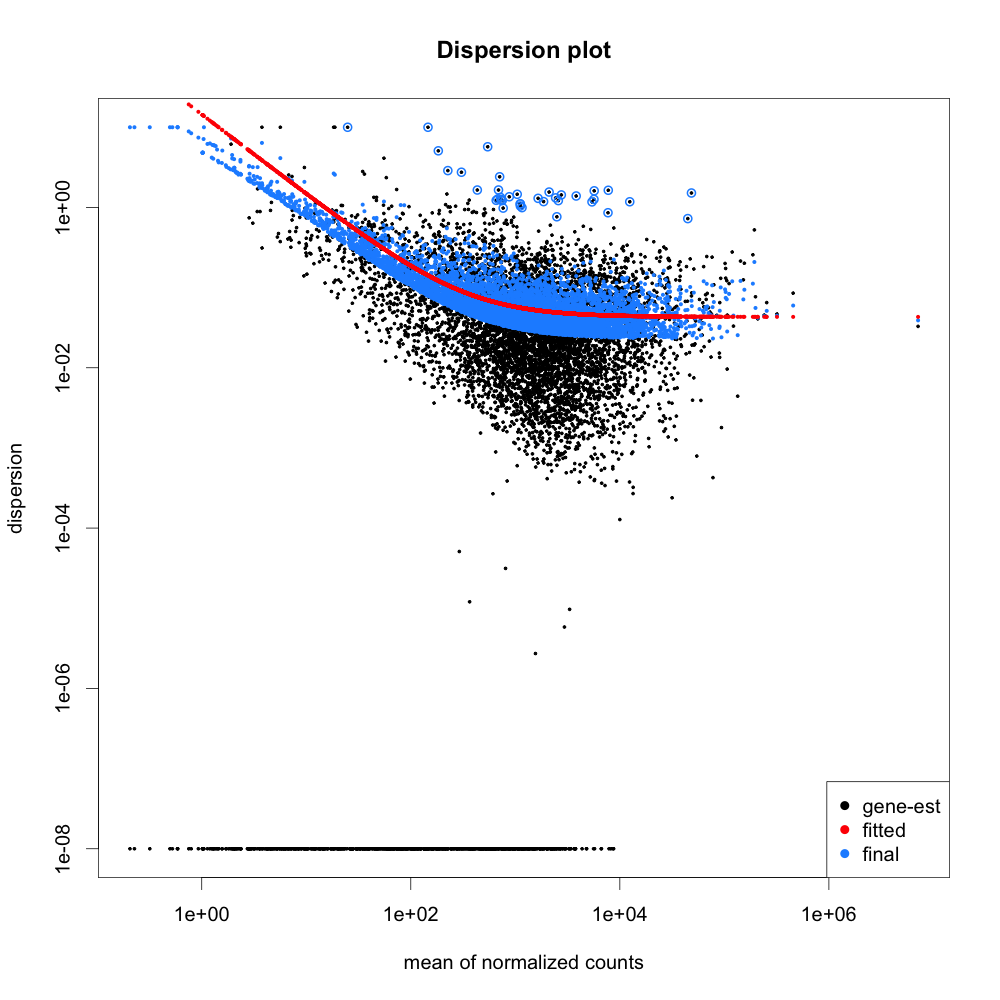
なんかイマイチなデータが多少含まれてそうだが、まぁ練習だし、良しとする。
作図
データ解析が終了したので、作図に移る。当初の目標通り、ヒートマップを作っていく。まずはデータをNormalized Counts Transformationする。
> ntd <- normTransform(dds)
そして、
> library("pheatmap")
> select <- order(rowMeans(counts(dds,normalized=TRUE)),
+ decreasing=TRUE)[1:20]
> df <- as.data.frame(colData(dds)[c("oxidate")])
> pheatmap(assay(ntd)[select,], cluster_rows=FALSE,
+ show_rownames=FALSE, cluster_cols=FALSE, annotation_col=df)
ただ、なんか画像のサイズがやけにデカイので、小さめにする。
> pheatmap(assay(ntd)[select,], cluster_rows=FALSE, show_rownames=FALSE,
+ cluster_cols=FALSE, annotation_col=df, cellheight=15,cellwidth=40)
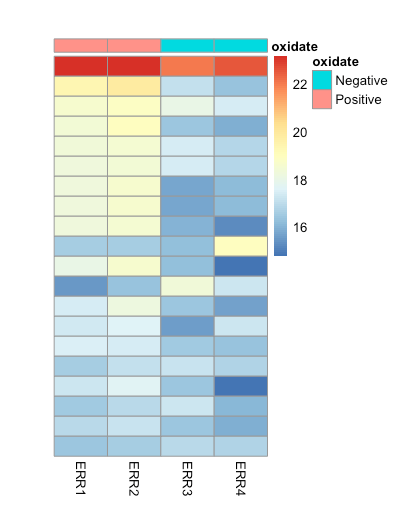
遺伝子名と系統樹みたいな線が欲しい時は以下のコマンドでできた。
> pheatmap(assay(ntd)[select,], cluster_rows=FALSE, show_rownames=TRUE,
+ cluster_cols=TRUE, annotation_col=df, cellheight=15,cellwidth=40)
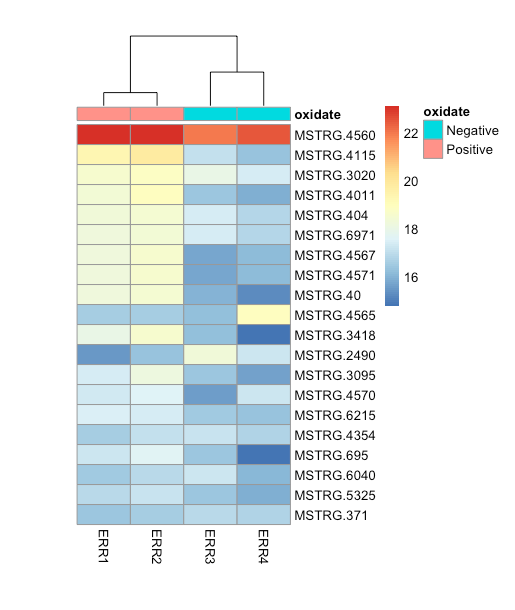
いくつかの遺伝子に変動がありそうだなという結果になった。ヒートマップは見やすいし、直感的にわかるので良いですね。
参考
StringTie: http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual
Github: https://gist.github.com/stephenturner/f60c1934405c127f09a6
余談
RNA-Seqデータ解析 Wetラボのための鉄板レシピを見ると、もっと詳しく色々書いてあります。RNA-Seqにおける発現解析で大事なGOタームの話とか書いてありました。自分のような素人にも大変分かりやすく載ってますし、今買えば、そこに載ってるコマンドで全て出来ます。非常に凄い本だと思いました。あんまり詳しく載ってなかったので、prepDE.pyについて書きました。