MA plotとは
MA plotは、一般的にはx軸を正規化した平均発現量、y軸を発現比とした散布図の一種で、主にRNA-seqやDNAマイクロアレイなどで得られた二群の遺伝子発現量を比較する際に用いられます。
数千、数万という遺伝子発現の変化を可視化することにより、重要な要素を特定する助けとなります。
例として、この図だと緑が統計的有意に発現量が増加した遺伝子で、赤が統計的有意に発現量が減少した遺伝子を示しています。
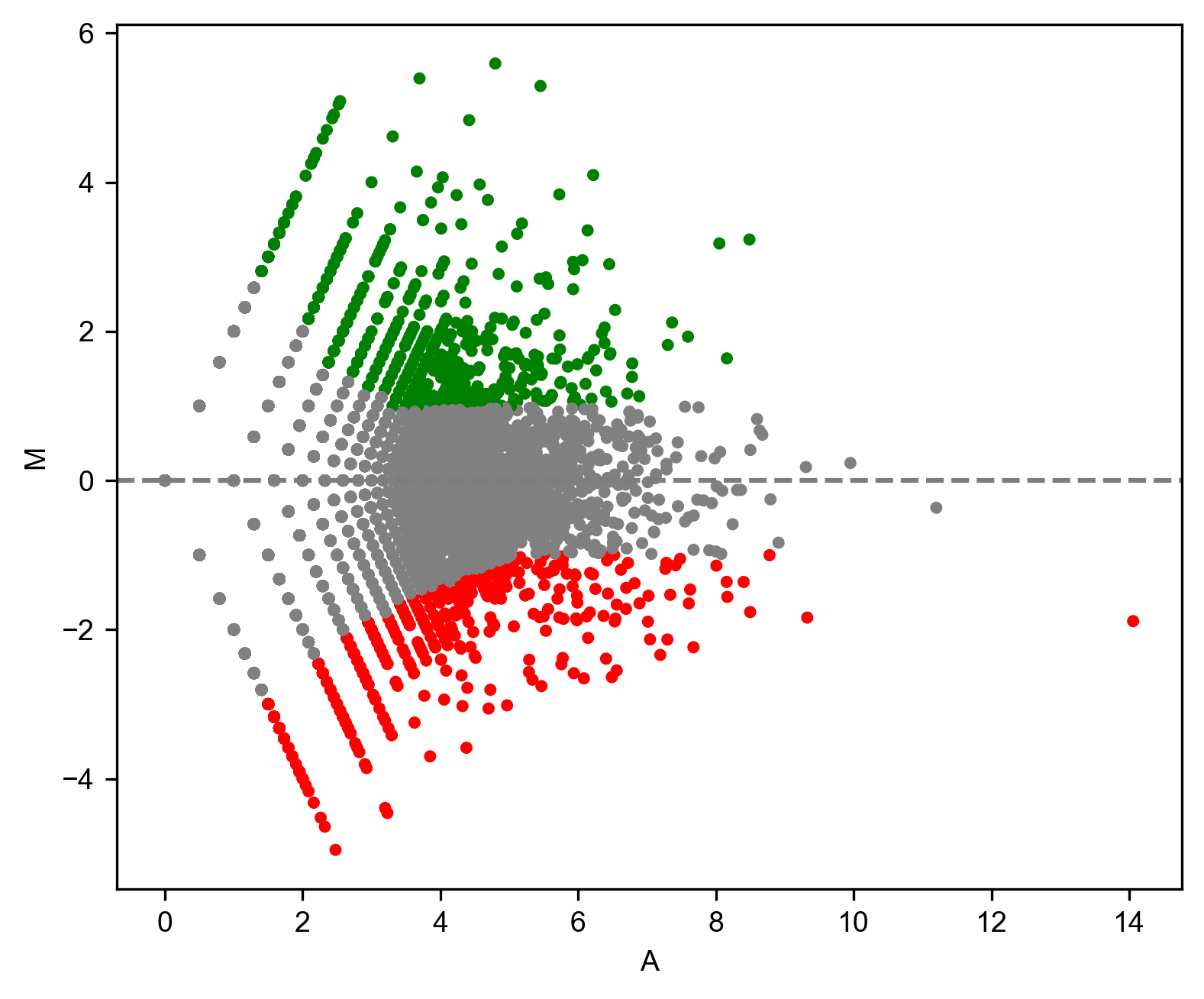
今回はこのMA plotをもっとカッコ良くしていこうと思います。
(pythonライブラリのbioinfokitのMA plot用データを使用)
完成図
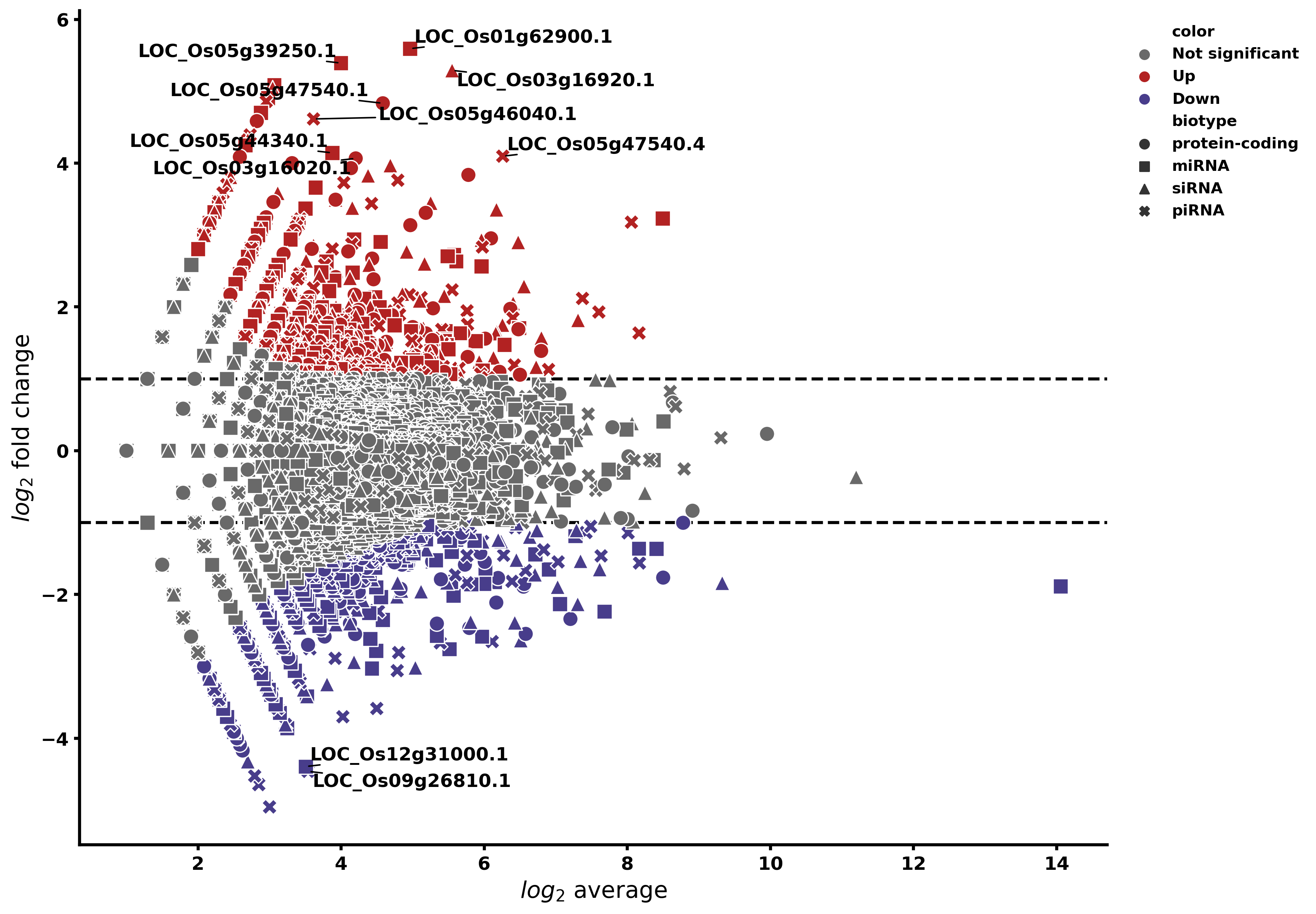
実践
必要なライブラリのインポート
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import numpy as np
from adjustText import adjust_text
import random#これは必須でない
from bioinfokit import analys, visuz
次にデータを読み込む。
df = analys.get_data('ma').data
df.head()
出力
GeneNames value1 value2 log2FC p-value
0 LOC_Os09g01000.1 8862 32767 -1.886539 1.250000e-55
1 LOC_Os12g42876.1 1099 117 3.231611 1.050000e-55
2 LOC_Os12g42884.2 797 88 3.179004 2.590000e-54
3 LOC_Os03g16920.1 274 7 5.290677 4.690000e-54
4 LOC_Os05g47540.4 308 18 4.096862 2.190000e-54
カラム名の名前を変更
df = df.rename(columns = {'p-value' : 'qvalue'})
df.head()
出力
GeneNames value1 value2 log2FC qvalue
0 LOC_Os09g01000.1 8862 32767 -1.886539 1.250000e-55
1 LOC_Os12g42876.1 1099 117 3.231611 1.050000e-55
2 LOC_Os12g42884.2 797 88 3.179004 2.590000e-54
3 LOC_Os03g16920.1 274 7 5.290677 4.690000e-54
4 LOC_Os05g47540.4 308 18 4.096862 2.190000e-54
新たなカラムを作成し、元データに各遺伝子の産物をランダムに追加
biotype = ['protein-coding', 'miRNA', 'siRNA', 'piRNA']
df['biotype'] = [random.choice(biotype) for i in range(len(df))]
df.head()
出力
GeneNames value1 value2 log2FC qvalue biotype
0 LOC_Os09g01000.1 8862 32767 -1.886539 1.250000e-55 siRNA
1 LOC_Os12g42876.1 1099 117 3.231611 1.050000e-55 piRNA
2 LOC_Os12g42884.2 797 88 3.179004 2.590000e-54 piRNA
3 LOC_Os03g16920.1 274 7 5.290677 4.690000e-54 siRNA
4 LOC_Os05g47540.4 308 18 4.096862 2.190000e-54 siRNA
次に、x軸用の値、平均発現量をbasemeanとして新たなカラムを作成する。
df['basemean'] = (np.log2(df['value1'] + 1) + np.log2(df['value2'] + 1)) / 2
df.head()
出力
GeneNames value1 value2 log2FC qvalue biotype basemean
0 LOC_Os09g01000.1 8862 32767 -1.886539 1.250000e-55 siRNA 14.056790
1 LOC_Os12g42876.1 1099 117 3.231611 1.050000e-55 piRNA 8.492965
2 LOC_Os12g42884.2 797 88 3.179004 2.590000e-54 piRNA 8.057989
3 LOC_Os03g16920.1 274 7 5.290677 4.690000e-54 siRNA 5.551644
4 LOC_Os05g47540.4 308 18 4.096862 2.190000e-54 siRNA 6.259695
ここまでで元データに必要なデータの追加が終了。
ここからはMA plotを描くためのデータ整形・加工になります。
MA plotの色分けのために新たなカラムを作成し、遺伝子発現量の差が統計的に有意であるものとそうでないものを分ける。
# MA plot必要なカラムのみを残す
df = df[['GeneNames', 'log2FC', 'qvalue', 'biotype', 'basemean']]
# 配色用の新たな関数の作成
def dot_color(a):
log2FC, qvalue = a
if abs(log2FC) <= 1 or qvalue >= 0.05:
return 'Not significant'#統計的に有意でないもの
if log2FC > 1 and qvalue < 0.05:
return 'Up'#有意に発現が上がったもの
if log2FC < -1 and qvalue < 0.05:
return 'Down'#有意に発現が下がったもの
# MA plotの配色用のカラムを作成
df['color'] = df[['log2FC', 'qvalue']].apply(dot_color, axis = 1)
df.head()
出力
GeneNames log2FC qvalue biotype basemean color
0 LOC_Os09g01000.1 -1.886539 1.250000e-55 siRNA 14.056790 Down
1 LOC_Os12g42876.1 3.231611 1.050000e-55 piRNA 8.492965 Up
2 LOC_Os12g42884.2 3.179004 2.590000e-54 piRNA 8.057989 Up
3 LOC_Os03g16920.1 5.290677 4.690000e-54 siRNA 5.551644 Up
4 LOC_Os05g47540.4 4.096862 2.190000e-54 siRNA 6.259695 Up
これでデータフレームの完成。
実際に使用してMA plotを描画してみる。
plt.figure(figsize = (12,10)) #サイズの指定
ax = sns.scatterplot(data = df, x = 'basemean', y = 'log2FC',
hue = 'color', hue_order = ['Not significant', 'Up', 'Down'],
palette = ['dimgrey', 'firebrick', 'darkslateblue'],
style = 'biotype', style_order = ['protein-coding', 'miRNA', 'siRNA','piRNA'],
markers = ['o', 's', '^', 'X'], s = 100)
# 統計的に有意な範囲を点線で区切る
ax.axhline(1, zorder = 0, c = 'k', lw = 2, ls = '--')
ax.axhline(-1, zorder = 0, c = 'k', lw = 2, ls = '--')
# 一定以上の統計的有意性を持つもののにannotationとして遺伝子名を表示させる
texts = []
for i in range(len(df)):
if df.iloc[i].basemean > 3.5 and abs(df.iloc[i].log2FC) > 4:
texts.append(plt.text(x = df.iloc[i].basemean, y = df.iloc[i].log2FC, s = df.iloc[i].GeneNames,
fontsize = 12, weight = 'bold'))
adjust_text(texts, arrowprops = dict(arrowstyle = '-', color = 'k'))
# 凡例の作成
plt.legend(loc = 1, bbox_to_anchor = (1.2,1), frameon = False, prop = {'weight':'bold'})
# 下と左の囲いを太くする
for axis in ['bottom', 'left']:
ax.spines[axis].set_linewidth(2)
# 上と右の囲いを消す
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.tick_params(width = 2)
plt.xticks(size = 12, weight = 'bold')
plt.yticks(size = 12, weight = 'bold')
plt.xlabel("$log_{2}$ average", size = 15)
plt.ylabel("$log_{2}$ fold change", size = 15)
# グラフの保存
plt.savefig('Qiita_maplot_test.png', dpi = 300, bbox_inches = 'tight')
plt.show()
出力
出来上がり!
どうでしょう...
少しごちゃごちゃしているような気もしますが、dotの形を新たな変数として設けたことで、個々の遺伝子の産物の種類までがわかるようになりました!
ちなみに、シンプルなMA plotでよければ、pythonライブラリのbioinfokitを使った方がもっと楽に作れます笑
なのでこのMA plotはカスタマイズ性とロマン重視になります笑