分子間相互作用計算
結晶中の分子間相互作用を計算したくなる時ってありますよね。CrystalExplorerというプログラムを用いると、分子間相互作用計算に関する知識があまり深くなくても有機結晶のcifファイルから、結晶中の分子間の相互作用を計算できます。しかも、CrystalExplorerではエネルギー分割計算もしてくれます。分子間相互作用と一言に言っても、分子間には電気的な相互作用である静電力($E_{ele}$)、永久双極子と誘起双極子の誘起力($E_{pol}$)、誘起双極子間に働くロンドン分散力($E_{dis}$)、反発項である交換反発力($E_{rep}$)をはじめとする様々な力が働いています。CrystalExplorerは分子間相互作用($E_{tot}$)を次のように計算してくれます。
$$
E_{tot} = k_{ele}E_{ele} + k_{pol}E_{pol} + k_{dis}E_{dis} + k_{rep}E_{rep}
$$
このように、分子間相互作用を個々の相互作用に分割してくれるのがエネルギー分割計算です。ここで、式の$k_{ele}$、$k_{pol}$、$k_{dis}$、$k_{rep}$はそれぞれ計算条件に依存する定数です。分子間相互作用のエネルギー分割をしてみて静電力の寄与が大きければ、たとえば、水素結合やハロゲン結合などの分子中の電荷が局在した箇所が分子全体の相互作用に効いているのかな、とか考えることができます。
CrystalExplorer
有機結晶中の原子間距離、二面角などを表示してくれたり、分子間相互作用を計算する解析プログラムです。Hirshfeld surface analysisという解析法を用いているとのこと。アカデミックの使用は無料です。Windows、MacOS、Linuxに対応しています。注釈の公式ホームページからダウンロードできます1。
ダウンロードしたCrystalExplorerを初めて開くときにはlicense codeを求められます。これも公式ホームページのLicensingで名前とメールアドレスを入力すると発行してもらえます。詳しいインストール方法はこちらを参照してください。
英語版ですが公式のwikiがあるので詳しい使い方はそちらを参照してください2。
インプットとアウトプット
・インプット
→結晶構造データ(.cif)
1)アスピリン3
インプットは結晶科学の分野でお馴染みcifファイルです。今回は薬効物質であるアスピリンの結晶をCrystallography Open Database(COD)4から無料でダウンロードして使います。ファイル名は1515581.cifでした。CODからダウンロードした結晶構造を以下に示します。
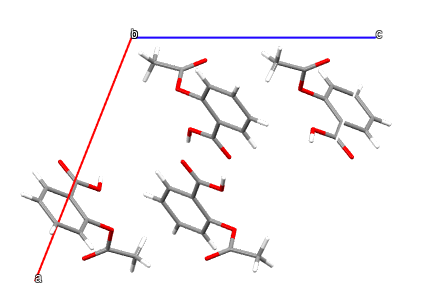
・アウトプット
計算結果はこんな感じに示されます。
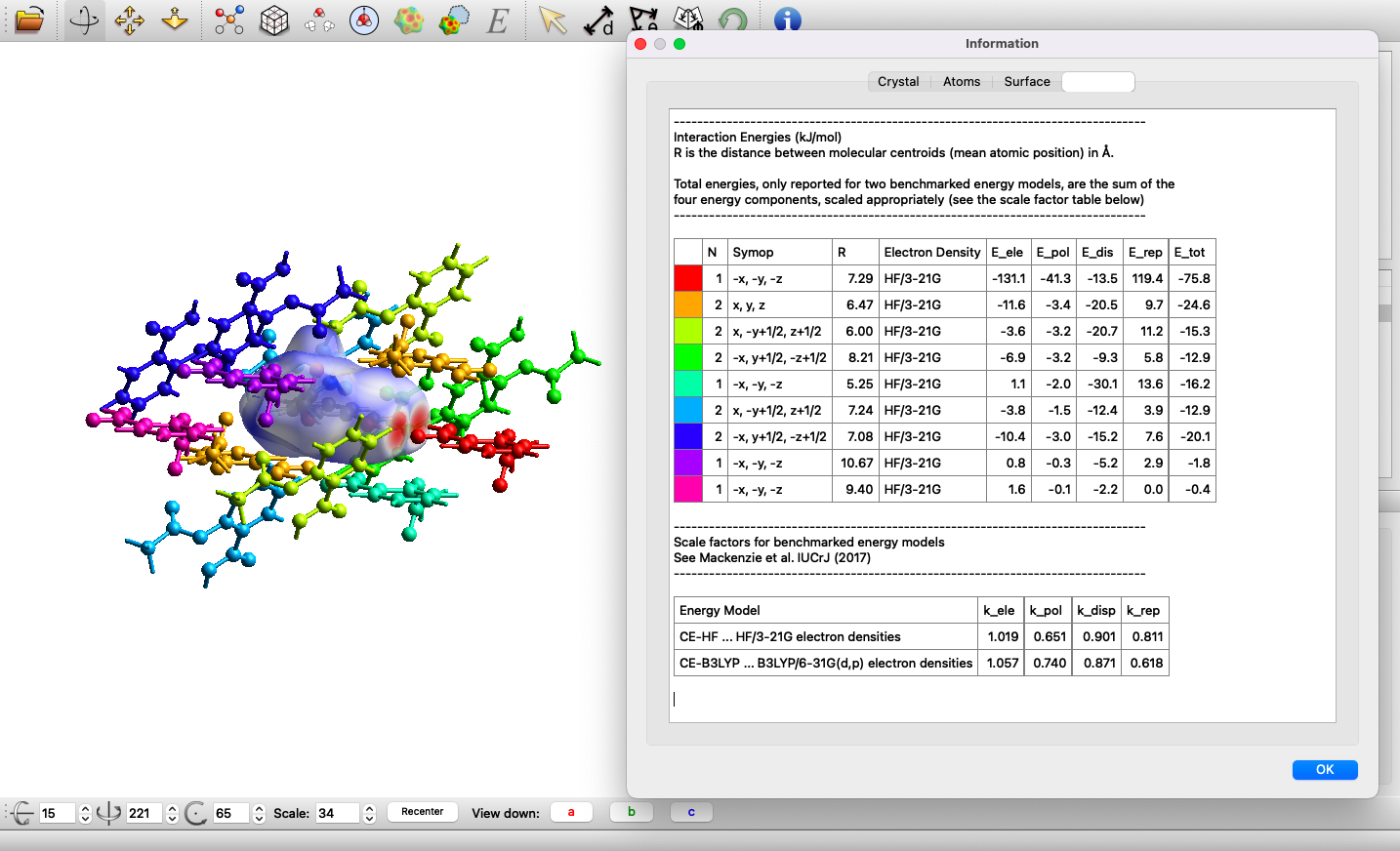
計算
1)ダウンロードしたCrystalExplorerを立ち上げます。
2)CrystalExplorerでアスピリン結晶のcifファイル(1515581.cif)を開きます。
3)右クリック-Select All Atomsで全原子選択します。選択された原子が黄色い網掛けになります。
4)Actions-Generate Surface...でHirshfeld surfaceを生成。出てくるポップアップはデフォルト設定のままOKでいいと思います。
5)再度、右クリック-Select All Atomsで全原子選択。
6)Actions-Calicurat Energys...を選択
7)1個目のポップアップはそのままOK。分子間相互作用を計算する分子が周辺に生成されます。
8)概ね次の図のような場面になると思います。計算方法を選択する箇所です。デフォルトはAccurateが選択されています。Accurateの方がFastより計算レベルが高いですが時間がかかります。今回の例では早く計算したいのでFastを選択してOKします。
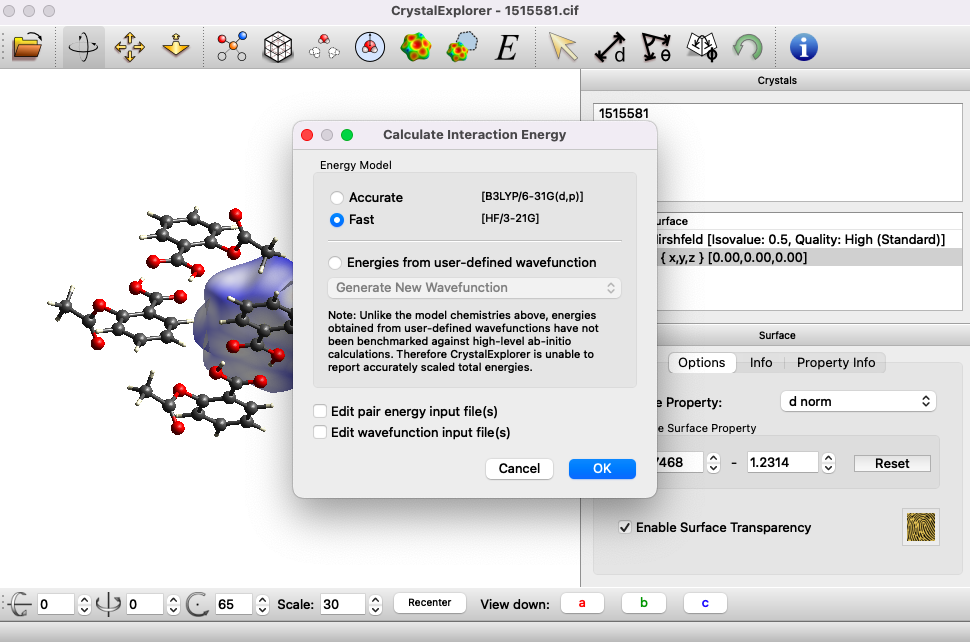
9)計算が終わったら計算結果が表示されます。
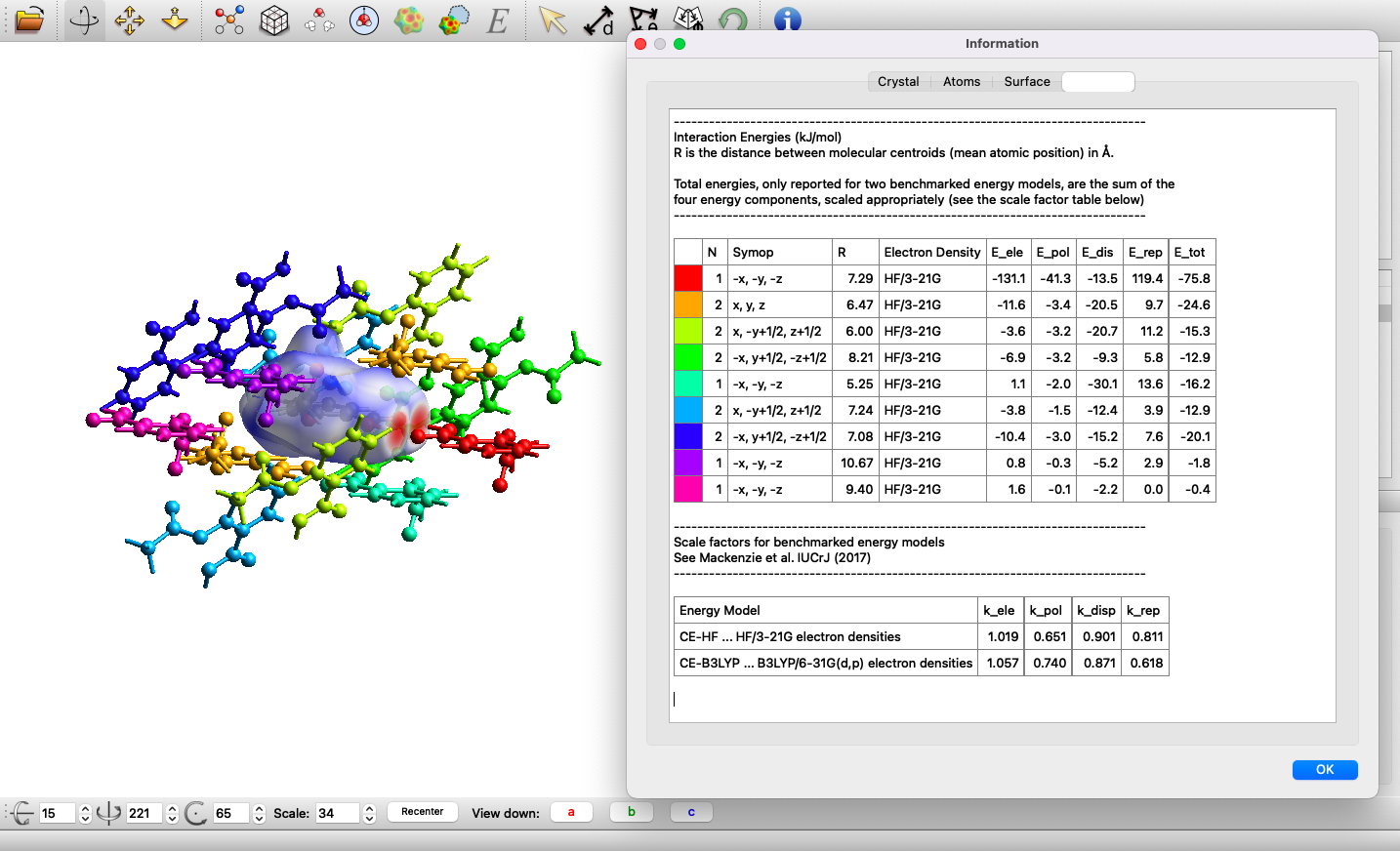
2)でcifファイルを開いたときに表示された分子と、7)で生成された周辺の分子との相互作用の値が表で示されています。表中のエネルギーの単位はkJ/molです。分子の色付けが表の左端の列の色と対応しており、Hirshfeld surfaceで囲われた中央の分子と赤い分子とのエネルギー分割計算の結果が一行目に書いてある値となります。
今回の計算結果から赤い分子との分子間相互作用が他の分子間相互作用に比べて十分大きく($E_{tot}=-75.8$ kJ/mol)、アスピリンは結晶中で二量体を形成していることが示唆されます。また、エネルギー分割計算の結果から赤い分子との分子間相互作用において静電力($E_{ele}=-131.1$ kJ/mol)の寄与が最大であり、その分子配置からも、アスピリンの二量体を形成にはカルボキシ基の水素結合の寄与が大きいことが推察されます。
表の二行目のオレンジの分子との間には、静電力($E_{ele}=-11.6$ kJ/mol)はあまり強く働かず、代わりに分散力($E_{dis}=-20.5$ kJ/mol)が強く働いていることがわかります。オレンジの分子よりも強い分散力が働いている分子もありますが、最も分散力が大きい分子は交換反発力も大きく、$E_{tot}$全体としては安定化がさほど大きくありません。電荷の偏りのない分子同士は近くにいる方が分散力が大きくなりますが、同時に交換反発力も大きくなります。結果的に、オレンジの分子との分子間相互作用が2番目に大きな相互作用($E_{tot}=-24.6$ kJ/mol)となっていますが、これはオレンジの分子が、適当な大きさの分散力を受けながら、適当な小ささの交換反発力のみが働くちょうどいい方位と距離にいるためであろうと考えられます。
-
CrystalExplorer公式
https://crystalexplorer.net/documentation ↩ -
CrystalExplorer wiki
https://wiki.crystalexplorer.net/ ↩ -
COD ID:1515581
アスピリンの結晶構造の文献。
Varughese, Sunil, et al. Chemical Science 2.11 (2011): 2236-2242. ↩ -
Crystallography Open Database
http://www.crystallography.net/cod/ ↩