先日発表されたDeepMind社のタンパク質立体構造予測AI「AlphaFold2」の論文についての解説です。
論文へのリンク
https://www.nature.com/articles/s41586-021-03819-2
技術的でない解説はコチラに書きました。
https://note.com/chanfuku18/n/n8f9613289bdb
問題設定
解きたいのは、タンパク質のアミノ酸配列から、立体構造座標を予測するという問題。アミノ酸配列という「暗号」を、立体構造に「翻訳」する問題とも捉えられています。ちなみにアミノ酸配列は、1次元の文字列データ。立体構造は、3次元×原子数分の実数データです。(アミノ酸は20種類あり、それぞれアルファベット1文字で表現されます。)

2年に1度、このタンパク質立体構造予測のコンテストが開催されており、2020年に圧倒的1位になったのがAlphaFold2。(2018年は、初代AlphaFoldが優勝。)
シーケンサー(配列を読む機械)の発達のおかげで、配列データは大量に入手できたのですが、実際にどういう形のタンパク質かは分かりませんでした。タンパク質を大腸菌などの細胞中で生成し、結晶解析で構造を調べる事もできますが、時間とお金がかかる為、計算機による予測が必要とされていました。
入力データ
物理化学の原理的には、アミノ酸配列のみから立体構造は予測できるはずなのですが、現状はそれは難しく、アミノ酸配列から得られる以下の情報も併せて特徴量として入力しています。
・Multiple Sequence Alignment: MSA
元のアミノ酸配列から類縁の似たようなアミノ酸配列をデータベースより収集して並べた(アライメントした)もの。専用のソフトウェアを用いて計算される。このアライメントの中には、生命の進化の情報が含まれると考えられ、構造予測の手がかりとなる。(立体構造上で近い位置にあるアミノ酸同士は、進化の過程で変異に統計的な相関がある事が知られており、AlphaFoldを含む予測モデルの多くは、この性質を利用している。)
・Template Structure
似たようなアミノ酸配列ですでに立体構造がわかっている場合、その構造情報を入力として用いている。(機械学習ではなく)そのような構造をテンプレートとして立体構造を予測する手法は20年以上前からあるが、AlphaFold2ではこれを特徴量として入力している。
基本戦略
機械学習を使わない方法も含め、これまで様々な予測手法が考えられてきたが、2016年あたりから上記のMSAをディープラーニングを用いて処理する方法が主流となっていた。
ただ、立体構造そのものを予測するのは難しいと考えられていた為、まずは、どの位置のアミノ酸同士が近い位置にあるかを予測するモデルが主流だった。(アミノ酸ペア毎の予測は、平面画像として表現できるので、CNNが使われてきた。)
しかし、出題されるアミノ配列は、数百~千以上のものが多く、立体構造予測では非局所的な相互作用が重要だと考えられる為、Attentionベースのモデルの方が良いと論文では主張している。
また、これまでは、アミノ酸ペア毎の予測距離を出力し、その距離を制約としてSimulated Annealing等により最適な立体構造を予測していたが、今回はEnd-to-Endで立体構造座標を出力している。End-to-Endで予測する手法はAlphaFold2が初めてという訳ではないが、あまり上手くいっている例がなかった。
モデル詳細
AlphaFold2のモデルは大きく分けると3つに分かれており、それぞれ、Embeddingパート、Evoformer パート、Structureパートとなっている。先に暗号の翻訳問題と書いたが、まさにAttentionを使った翻訳モデルのような構造をしている。
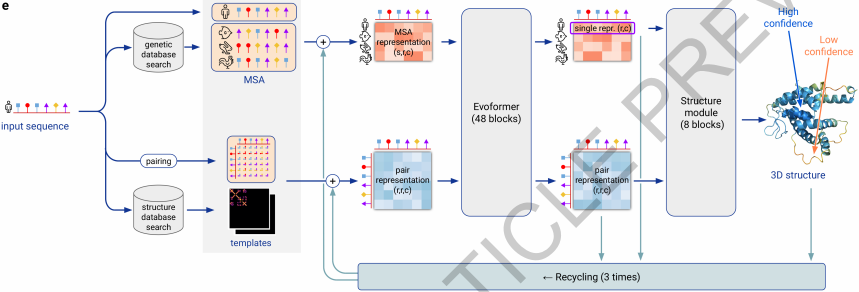
Embeddingパート
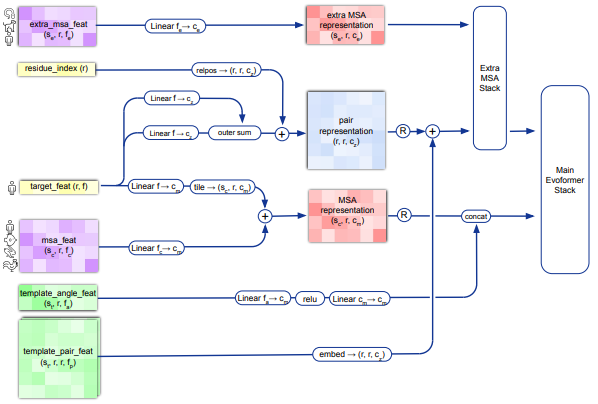
単純に入力された特徴量をMLP等で次元圧縮している部分。最終的に、
MSA representation: アミノ酸配列の長さ×類縁配列のサンプリング本数×256次元
pair representation: アミノ酸配列の長さ×アミノ酸の長さ×128次元
としてEvoformerパートへ出力している。(最後の次元は画像におけるチャンネル数に相当)
Evoformerパート
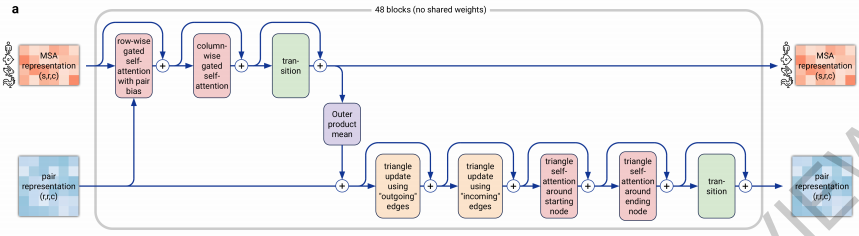
入力された特徴量をAttentionを用いてEncodingしている部分。
(EvoformerはEvolution+Transformerからの造語と思われる。)
同じblockを48回繰り返し、最終的に、
pair representation: アミノ酸配列の長さ×アミノ酸の長さ×128次元
single representation: アミノ酸配列の長さ×384次元
としてStructureパートに出力している。
Evoformer blockは、MSA stack(図上段)とpair stack(図下段)、およびそれらのcommunication部(図中段)から構成されている。
入力されるMSA representationもpair representationは画像と同じ次元を持っている為、Attentionも画像処理と同様に処理されており、Huang+ 19およびWang+ 20で提案されたaxial attentionを用いている。
また、三角形の二辺の和が残りの一辺より大きいという制約を満たす様に、Pair stack内では、3点の情報を元にattentionを計算するtriangular self-attentionという手法を開発している。
Structureパート
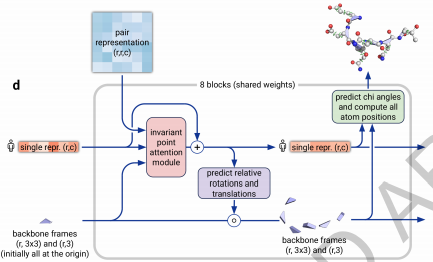
Evoformerでencodingされた特徴量をdecodingしている部分。
8回繰り返しているが、その度に立体構造を時系列として出力する。タンパク質の構造はアミノ酸が直鎖上につながっており、1つのアミノ酸毎に、3つの角度で立体構造を表現している。(スネークキューブのようなものをイメージしてください。)
これまで2つのアミノ酸間の距離を出力する(後工程でその距離制約に基づいてsimulated annealingを走らせる)のが主流だったのに対して、立体構造をend-to-endで出力するのがポイント。
ここで、立体構造の扱い方に工夫があり、各アミノ酸の中心原子(一般にCα原子と呼ぶ)を中心とした座標系(図中のbackbone frame)を導入。各アミノ酸のCα原子の位置で大まかな構造を表現し、アミノ酸内の詳細な構造はそこからの相対位置で計算する。
また、3次元構造内でattentionを行う工夫として、invariant point attention(IPA)という手法を開発している。
訓練方法
・1000万sampleを、128個のTPU v3で、1週間かけて訓練。さらにfine-tuningに4日間。
(TPUが1時間$8として約$17万。約2000万円弱。)
・Frame aligned point error(FAPE) lossという損失関数を用いて最適化。
式は下記の通りだが、基本的には距離を最小化している。
(実際は、FAPE lossを含め、合計7つの損失関数が用いられている。)

その他の工夫など
・Self-distillationを用いたモデルも作成。
・最終構造はAmberエネルギー関数を用いて最適化しているが、scoreには寄与しない。
・モデルの信頼度をpLDDTという値で出力する事ができる。
・16GBのGPUメモリでは、アミノ酸配列の長さ1300が上限。
考察
実は、AlphaFold2と言えども、100%完璧な予測ができる訳ではなく、
・見つかっている類縁配列が少なく、進化の情報が十分でない場合。
・近接しているアミノ酸ペアが少ない構造の場合。(アミノ酸間の距離情報が乏しい為。)
には予測精度が落ちると論文では述べられている。
個人的雑感等
・本論文は、「ディープラーニングの最新の知見をふんだんに盛り込んだモデル」と「圧倒的な計算機リソース」というDeepMindならではの強みが生きている。(ただ、AlphaGoの様な強化学習がメインのモデルではない。)
・初代AlphaFoldの時は、あまり論文に詳細が出ていなかったが、今回はsupplementで62ページに渡ってモデル詳細に説明し、実装も公開している。「タンパク質フォールディング問題」を解いたという科学的な意味合いに対して、再現性を担保する必要があったのかもしれない。ディープラーニングが科学の発展に大きく貢献した最初の例になるかもしれない。
・ディープラーニングのモデルは、MLPをベースに、CNNやRNNが開発され、attentionが登場し、最近改めてMLPに脚光が浴びたりしながら、発展が続いている。よくinductive biasと表現されているが、「ドメイン知識に基づいてネットワーク構造を規定した事が今回の勝因」と、first authorであるJumperさんが講演でコメントしている。
・初代AlphaFoldも含めて、多くの研究者がアミノ酸の距離情報の予測と、その後工程としての最適化を別々に扱っていたが、AlphaFold2ではEnd-to-Endでこれを実現した。End-to-Endで学習することで、最適化計算に相当する部分が省略できているのが興味深い。(試したことはあったが、全く上手く行く気配がなかった…。)
・アカデミック界で長年この領域を牽引してきたDavid Bakerさんも、コンテストでは敗れたものの今回の同時期に新しい論文を発表し、意地を見せている。