MatterSimを使って材料シミュレーション!物性予測・フォノン・MD計算まで一気通貫でやってみた【Python/ASE】
こんにちは。材料シミュレーションやマテリアルズインフォマティクス(MI)に関心のある方向けに、Microsoftが公開している軽量な第一原理シミュレータ MatterSim を使うための環境構築と簡易的な解析ををご紹介します。
🔍 はじめに 〜MatterSimとは何か、なぜ使うのか〜
第一原理計算(例:DFT)による材料の物性解析は高精度ですが、計算コストが非常に高いため、材料スクリーニングや長時間スケールの現象(フォノン・MD)には不向きです。
そこで注目されているのが 機械学習ポテンシャル(MLP) です。これは、第一原理計算で得たエネルギーや力のデータを教師として学習し、同等の精度をより高速に再現するものです。
MatterSimは、Microsoftが開発した 軽量かつ高精度なMLP であり、以下のような特徴があります:
- M3GNetをベースに構築されており、グラフニューラルネットワーク(GNN)を活用したアーキテクチャ
- 多様な材料データをアクティブラーニングで統合して学習済み
- 結晶系の様々な構造に対応
- 軽量なPythonモジュールとして提供され、ASEとの親和性も高く、初学者〜研究者まで手軽に使える
📊 ベンチマーク精度(参考:公式論文より)
以下は、MatterSimの開発論文[^1]に掲載された主要な機械学習ポテンシャルとの比較ベンチマークです。予測対象は、構造安定性に関わる分類およびエネルギー回帰で、性能指標として F1スコア, 精度 (Accuracy), MAE, RMSE, 決定係数 R² などが使われています。
| モデル | F1 | Precision | Accuracy | MAE | RMSE | R² |
|---|---|---|---|---|---|---|
| MatterSim | 0.83 | 0.83 | 0.96 | 0.03 | 0.08 | 0.81 |
| GNoMe | 0.81 | 0.83 | 0.94 | 0.03 | 0.08 | 0.78 |
| CHGNet | 0.58 | 0.52 | 0.84 | 0.07 | 0.11 | 0.61 |
| M3GNet | 0.57 | 0.45 | 0.80 | 0.07 | 0.11 | 0.60 |
| MACE | 0.57 | 0.47 | 0.81 | 0.07 | 0.11 | 0.63 |
| ALIGNN | 0.56 | 0.50 | 0.83 | 0.09 | 0.15 | 0.27 |
| MEGNet | 0.51 | 0.46 | 0.81 | 0.13 | 0.20 | -0.28 |
| CGCNN | 0.51 | 0.45 | 0.81 | 0.14 | 0.23 | -0.62 |
| Wrenformer | 0.48 | 0.36 | 0.74 | 0.10 | 0.18 | -0.04 |
| BOWSR | 0.44 | 0.32 | 0.68 | 0.12 | 0.16 | 0.14 |
→ MatterSimはほぼすべての指標で他モデルを上回る性能を示しており、特に分類(F1)と回帰(R²)の両方で高い性能を発揮していることが分かります。
📄 出典:Zhao et al., MatterSim: A general-purpose atomistic machine learning simulator, arXiv:2405.04967
このように、MatterSimは軽量ながら高精度なMLPとして、材料研究やマテリアルズインフォマティクス(MI)分野で非常に有用なツールです。
🧠 1. MatterSimの原理 〜M3GNetからの発展とモデル構造〜
MatterSimは、M3GNet(Materials Graph Networks) をベースに構築されたグラフニューラルネットワーク(GNN)モデルです。M3GNetは、材料の構造をグラフとして表現し、原子間の関係性をベクトルとして学習できる点が特徴です。
MatterSimではこのM3GNetの基本構造に加え、
- 🔁 アクティブラーニング(Active Learning) により、さまざまな材料構造を反復的に取り込みながら精度を向上
- 🧩 複数のデータセット(MP-DOS、JARVIS、OC20など)を統合し、多様な系に対応
- ⚙️ 力・応力・エネルギーを同時に予測可能な多出力モデル
として拡張されています。
その結果、汎用的で高精度なエネルギーサーフェスを再現できるようになっており、従来のポテンシャル(Stillinger-Weber, Tersoffなど)では困難だった未知系や複雑な構造に対しても柔軟に対応可能です。
⚙️ 2. 環境構築 〜PythonとMatterSimの導入〜
🔹 前提:conda環境がインストールされていること
MatterSimはPythonベースのツールであり、パッケージの依存関係も多いため、conda を使った仮想環境での導入を推奨します。
Minicondaをまだ導入していない方は、以下からインストールしてください:
👉 https://docs.conda.io/en/latest/miniconda.html
2.1 仮想環境の作成とアクティベート
conda create -n mattersim python=3.9
conda activate mattersim
2.2 必要なライブラリのインストール
コピーする
# MatterSim(GitHubから最新を取得)
pip install git+https://github.com/microsoft/mattersim.git
# 可視化・構造処理に必要なライブラリ
pip install ase matplotlib
ase:Atomic Simulation Environment。構造生成やMD、フォノン解析に使用
matplotlib:フォノン分散などの可視化に使用
🖥️3. 構造作成と物性予測 〜Si, NaCl, MgAl₂O₄を例に〜
このセクションでは、以下の3つの代表的な無機材料を対象に、構造作成および構造緩和とMatterSimによる物性予測(エネルギー・力)を行います:
| 材料 | 構造 |
|---|---|
| Si | ダイヤモンド構造 |
| NaCl | 岩塩構造 |
| MgAl₂O₄ | スピネル構造 |
構造生成にはすべて ASE を用い、MgAl₂O₄については Materials Project mp-3536 を参照して格子定数・原子配置を明示的に指定します。
3.1 構造の作成と計算の実行
from ase import Atoms
from ase.build import bulk
from ase.optimize import BFGS
from ase.filters import FrechetCellFilter
from mattersim.forcefield import MatterSimCalculator
# 結晶Si、NaCl
atoms_si = bulk('Si', 'diamond', a=5.43)
atoms_nacl = bulk('NaCl', 'rocksalt', a=5.64)
# MgAl2O4(mp-3536準拠)
a = 8.083
positions = [
(0.125, 0.125, 0.125), # Mg
(0.5, 0.5, 0.5), (0.0, 0.5, 0.5), (0.5, 0.0, 0.5), (0.5, 0.5, 0.0), # Al
(0.260, 0.260, 0.260), (0.740, 0.740, 0.740),
(0.740, 0.260, 0.260), (0.260, 0.740, 0.260), (0.260, 0.260, 0.740),
(0.740, 0.740, 0.260), (0.260, 0.740, 0.740), (0.740, 0.260, 0.740)
]
symbols = ['Mg'] + ['Al'] * 4 + ['O'] * 8
atoms_mgal2o4 = Atoms(symbols=symbols,
scaled_positions=positions,
cell=[(a, 0, 0), (0, a, 0), (0, 0, a)],
pbc=True)
# MatterSim計算器
calc = MatterSimCalculator()
# 最適化&出力
results = []
for atoms, name in zip([atoms_si, atoms_nacl, atoms_mgal2o4], ['Si', 'NaCl', 'MgAl2O4']):
atoms.calc = calc
# セル+構造緩和(推奨フィルタ)
fcf = FrechetCellFilter(atoms)
opt = BFGS(fcf, logfile=f"{name}_opt.log")
opt.run(fmax=0.01)
# 結果取得
energy = atoms.get_potential_energy()
forces = atoms.get_forces()
max_force = forces.max()
cell = atoms.cell.cellpar()
# 構造ファイル保存(CIF形式)
atoms.write(f"{name}_relaxed.cif")
# 出力
results.append((name, energy, max_force, cell))
print(f"===== {name} =====")
print(f"Energy: {energy:.4f} eV")
print(f"Max Force: {max_force:.4f} eV/Å")
print(f"Cell: {cell}")
print(f"Structure saved to: {name}_relaxed.cif\n")
上記を実行した結果は以下の通りです。
$ python test.py
2025-04-19 22:58:34.275 | INFO | mattersim.forcefield.potential:from_checkpoint:877 - Loading the pre-trained mattersim-v1.0.0-1M.pth model
===== Si =====
Energy: -10.8291 eV
Max Force: 0.0000 eV/Å
Cell: [ 3.86321225 3.86321226 3.86321229 60.00000312 60.00000159 60.00000155]
Structure saved to: Si_relaxed.cif
===== NaCl =====
Energy: -6.7816 eV
Max Force: 0.0000 eV/Å
Cell: [ 4.02788192 4.0278819 4.02788189 60.00000031 59.99999996 60.00000005]
Structure saved to: NaCl_relaxed.cif
===== MgAl2O4 =====
Energy: -89.1118 eV
Max Force: 0.0123 eV/Å
Cell: [ 5.83261055 4.8721698 4.87045268 102.97614428 89.98007399
89.99520343]
Structure saved to: MgAl2O4_relaxed.cif
3.2 結果と考察(DFTとの比較)
以下に、MatterSimの出力結果と、Materials Projectに基づくDFTの比較をまとめます:
| 材料 | MatterSimエネルギー (eV) | Max Force (eV/Å) | DFTエネルギー (eV) | 備考・参照 |
|---|---|---|---|---|
| Si | -10.8291 |
0.000 |
-17.548 |
mp-149 |
| NaCl | -6.7816 |
0.000 |
-13.793 |
mp-22862 |
| MgAl₂O₄ | -89.1119 |
0.013 |
-105.007 |
mp-3536 |
※ エネルギーはすべてユニットセルあたり。Max Force は原子ごとの最大原子間力。
⚠️ エネルギーの比較についての注意
MatterSimとDFTで得られる構造全体のエネルギー値(絶対値)には大きな差が見られます(例:MgAl₂O₄で15 eV以上)。これは次のような理由によるものであり、直接の一致を期待するものではありません:
- MatterSimはエネルギー差(相対安定性)に焦点を当てて学習されており、絶対値はゼロ点シフトやバイアスを含む可能性がある
- DFTのように明示的にゼロ点エネルギーを規定していない
👉 よって、MatterSimの評価には、以下のような観点が重要です:
- 構造ごとのエネルギー差の傾向
- 構造緩和後の力の大きさ(静的安定性)
- フォノン分散など微分物理量の一致性
本記事では、フォノンバンド構造に着目して、MatterSimの有効性を検証します。
🚀4. Phonon計算と機械学習ポテンシャルの評価
4.1 Phononとは?
Phonon(フォノン)は、結晶格子中の原子の集団的な振動(格子振動)を量子化して表現した準粒子です。フォノンの性質は、以下のような多くの物性に影響を与えます:
- 比熱、熱伝導率
- 音速、弾性定数
- 電子—フォノン相互作用による超伝導
これらの性質は、フォノンの**分散関係(バンド構造)**を解析することで理解できます。
🔬 4.2 なぜPhonon計算が機械学習ポテンシャルの評価に有効なのか?
Phononのバンド構造を求めるには、力定数(force constants)、すなわち原子を微小に変位させたときに働く二階微分ポテンシャルが必要です。
つまり:
Phonon計算は、力の精度を極めて敏感に評価する方法である。
そのため、MLポテンシャル(MatterSimなど)を使ったシミュレーションで「どこまで第一原理計算に近い力を再現できているか?」を評価するには、Phononバンド構造の比較が非常に有効です。
4.3 複数構造(Si、NaCl、MgAl₂O₄)に対するPhononバンド構造の計算
以下のスクリプトでは、**前節で緩和した3つの構造ファイル(*_relaxed.cif)**を読み込んで、Phononバンド構造を一括で計算・プロットします。出力は自動的に各構造ごとに分けて保存されます
from ase.io import read
from mattersim.forcefield import MatterSimCalculator
from mattersim.applications.phonon import PhononWorkflow
from ase.dft.kpoints import bandpath
import numpy as np
import os
systems = [
("Si_relaxed.cif", "Si", (3, 3, 3), ['G', 'X', 'W', 'K', 'G', 'L']),
("NaCl_relaxed.cif", "NaCl", (3, 3, 3), ['G', 'X', 'W', 'K', 'G', 'L']),
("MgAl2O4_relaxed.cif", "MgAl2O4", (2, 2, 2), ['G', 'X', 'W', 'K', 'G', 'L'])
]
for cif, label, sc, kpath in systems:
print(f"\n=== Phonon計算開始: {label} ===")
# CIFファイルの読み込み
atoms = read(cif)
# 計算機を割り当て
atoms.calc = MatterSimCalculator()
# 作業ディレクトリの設定
work_dir = f"./phonon_{label}"
os.makedirs(work_dir, exist_ok=True)
# フォノンワークフローの構築
ph = PhononWorkflow(
atoms=atoms,
find_prim=False,
work_dir=work_dir,
amplitude=0.01,
supercell_matrix=np.diag(sc)
)
# 実行
has_imag, phonons = ph.run()
print(f"Has imaginary phonon: {has_imag}")
🎯 4.4 結果の比較と考察
以下に、各構造で計算されたPhononバンド構造をまとめ、特徴や精度について評価します:
Si

全体として、第一原理計算の結果と良く一致しており、基本的な振る舞いが適切に再現されていることがわかります。
NaCl
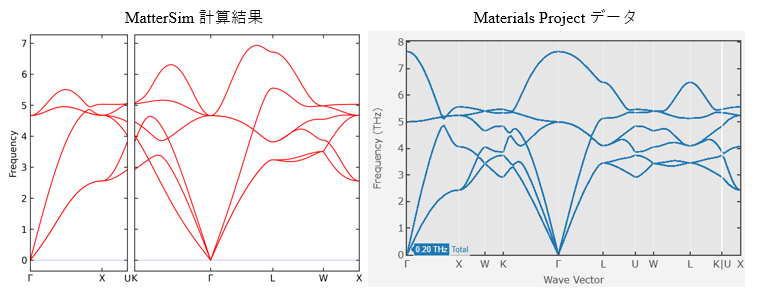
分散の形状は概ね一致しているものの、LO-TO分裂はMatterSimがBORN補正を含まないため再現されていません。
NaCl

Materials Project に該当データが存在しないため、MatterSimによる計算結果のみを示しています。複雑な構造であっても短時間でPhonon特性を得られる点は特筆に値します。
👀 観察された傾向:
-
**低周波数領域(音響モード)**は非常に高い精度で再現されており、結晶構造の安定性評価には十分に有効です
-
一方で、**高周波数領域(光学モード)**では、周波数のずれ、過剰なバンド分裂、非物理的な平坦化が一部で見られました。
-
LO-TO分裂は、前述の通りBORN補正が未実装のため再現されません。
4.5 まとめ:Phonon計算を通じたMatterSimの有効性
本章では、Phonon分散関係を通じて、MatterSimがどの程度第一原理計算に近い力場を再現できるかを検証しました。
📌精度面の評価
構造緩和後の力の大きさは 0.01 eV/Å 未満で十分静的平衡に到達。
フォノンの分散傾向は良好であり、材料設計の初期評価に使える精度。
絶対周波数や高精度な熱物性評価には、第一原理との併用が望まれる。
⚡計算効率
従来のDFTベースのPhonon計算と比較して数百倍の高速化が可能。
MgAl₂O₄のような複雑構造でも10分以内に計算が終了。
環境構築が比較的容易で、SIMによるスクリーニング技術として非常に有用。
✅結論:
MatterSimは、Phononバンドを用いた力場評価において、
初期設計・スクリーニング用途において非常に有効なツールです。
完全に第一原理を代替するものではないものの、
精度とスピードのバランスが極めて良好であり、
研究開発現場での導入価値が高いと評価できます。
🧩おわりに
MatterSimは、第一原理計算の代替となる軽量な物理モデルとして、
マテリアルズ・インフォマティクス(MI)やAIを活用した材料研究において、
⏱️ 高速スクリーニングや
🧪 初期探索にとても役立つツールです。
💡 材料系×AIで気軽にシミュレーションしてみたい方におすすめ!
材料系×AIで手軽にシミュレーションしてみたい方におすすめです!
参考資料
- MatterSim GitHub
- MatterSim ドキュメント
- arXiv論文(2024): MatterSim: A general-purpose ML potential with phonon and thermodynamic accuracy
- ASE 公式(Atomic Simulation Environment)
🙌 もし参考になったら「いいね」や「ストック」お願いします!
X(Twitter): @ap123hao (フォロー歓迎です)