お久しぶりです。ということで続けていきましょう!
今回は、前回行ったタンパク質相互作用のデータをPGXに読み込ませるように出来た続きとして、さらにこのデータ内に薬剤とタンパク質のつながりのデータを加えて(統合させて)みようと思います!!![]()
すること
①タンパク質と薬剤のつながりのデータを入手する
このデータを入手するために、今回は「DrugBank」を使用しました!
DrugBankとは、薬剤自体のデータ(構造式、機能など)や、薬剤が結合、作用するもの(タンパク質など)の情報(何と何が結合しているのか?どこに結合しているのか?など)を提供しているサイトです。
ただし、データを入手するにはアカウント登録が必要です!!
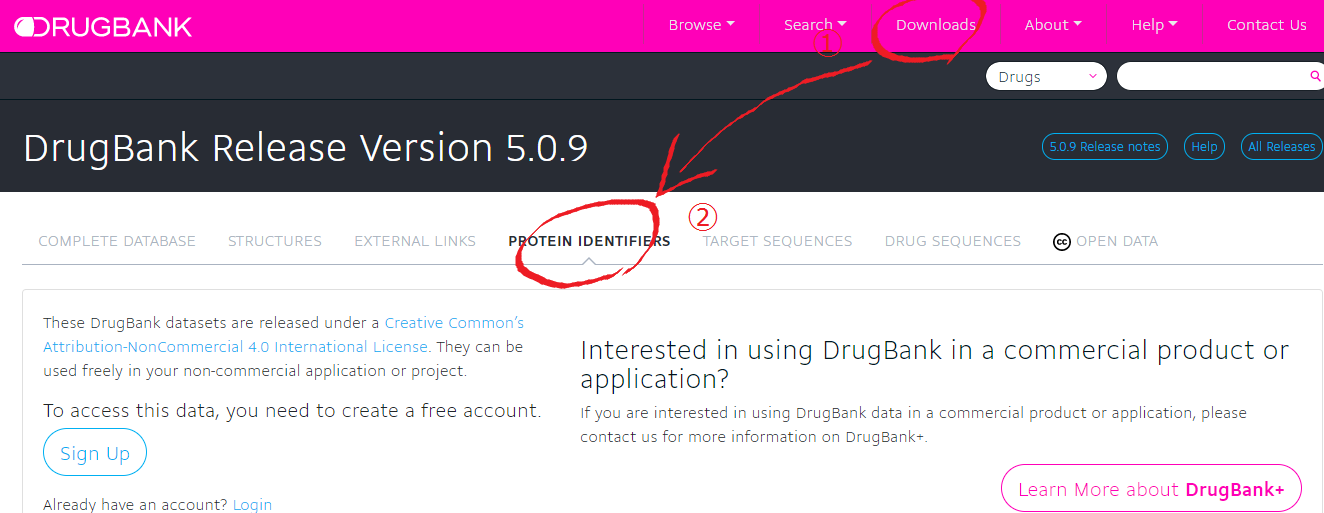
入手したい情報は**どの薬剤とどのタンパク質が結合しているのか?**なので、「DrugBank」のページ内にある<Downloads⇒PROTEIN IDENTIFIERS>の順に選択し、そのページの<Drug Target Identifiers>の欄のAllをダウンロードします。
$ curl -Lfv -o filename.zip -u EMAIL:PASSWORD https://www.drugbank.ca/releases/5-0-9/downloads/target-all-polypeptide-ids
☝実際にサイト内に掲載されているコマンドです!コマンドのEMAIL:PASSWORDにはアカウント登録に用いたEMAIL、PASSWORDを入力してください。もちろんGUIの操作でローカル環境に落とすこともできます!(一番右側の<DOWNLOAD(CSV)>を選択)
ダウンロードしたデータはzip形式で保存されます。その後、解凍すると2つのcsvファイルが出来ます。今回はこのうちのall.csvという方を用います!(実際にそのつながりの意味の有無に関わらず結合するデータが全て入っているものです)
このデータと、前回の記事で作成したデータを用いて行っていきます!
②必要な情報を抽出して整形する
# !/bin/bash
sed 's/"\([^"]*\)"//g' all.csv > temp_all_1.csv
awk -v FS=',' -v OFS=',' '{if (NR != 1) print $1, $3}' temp_all_1.csv > temp_all.csv
awk -v FS='\t' -v OFS=',' '{if (NR != 1) print $1, $3}' uniprot.tab > temp_uniprot_id.csv
awk -v FS=',' -v OFS=',' '{if (NR != 1) print $1, $13}' temp_all_1.csv > temp_drug_ids.csv
awk -v FS=',' -v OFS=',' '{split($2, array, "; "); for (i in array) { print $1, array[i] }}' temp_drug_ids.csv > temp_drug_ids_split.csv
sort -t ',' -k 2 temp_uniprot_id.csv | uniq > temp_uniprot_id_sorted.csv
sort -t ',' -k 1 temp_drug_ids_split.csv > temp_drug_ids_sorted.csv
join -t ',' -1 2 -2 1 -o 0 1.1 2.2 temp_uniprot_id_sorted.csv temp_drug_ids_sorted.csv > temp_drug_edge.csv
awk -v FS=',' -v OFS=',' '{print $2,$3=$3",1"}' temp_drug_edge.csv > drug_edge.csv
awk -v FS=',' -v OFS=',' '{print $2,$1,"0","0","0","0","0","0","0","0"}' drug_edge.csv | sort | uniq > detailed_drug_edge.csv
rm temp_* drug_edge.csv
こちらはグラフのエッジにあたるデータです。前回と異なる点として、
- データ内でのカンマ区切り以外にタンパク質名等の中にカンマがあるなどしてうまく認識してもらえなかったので、最初にカラムごとの完全なカンマ区切りにしたこと
- Drug IDに関するカラムで、あるタンパク質に結合する薬剤が1つ以上である、つまりそのカラム内で「;」 区切りで薬剤IDが記述されていたので、それをバラバラにさせて一つ一つに分けた(forループ構文を用いている)こと
例:Protein ID,…,DB00023; DB00044; DB00099 を
Protein ID,DB00023
Protein ID,DB00044
Protein ID,DB00099 にする
- エッジのプロパティは値がないが、前回作成したエッジプロパティの数と合わせる必要があるため、その部分に「0」の値を挿入したこと
があります。
# !/bin/bash
awk -v FS=',' -v OFS=',' '{print $1,"*",$1}' detailed_drug_edge.csv | sort | uniq > drug_node.csv
こちらはノードに当たるデータの抽出です。ノードプロパティにはID番号を入れています。
そしてこれらのデータと前回作成したPPI(タンパク質相互作用)のデータとを統合させます。
# !/bin/bash
sh create_ppi_node.sh 100000
sh create_ppi_edge.sh 12000000
sh create_drug_edge.sh
sh create_drug_node.sh
cat drug_node.csv detailed_drug_edge.csv ppi_node.csv ppi_edge.csv > drug_ppi_edgelist.csv
コマンド内の数字は引数です。適宜ちょうどいい引数を入れてください!
③PGXに読み込ませる
前からずーっと言っていますが、PGXに読み込ませるためにはJSONファイルが必要になります。
{
"uri": "置いてあるディレクトリまでのパス/drug_ppi_edgelist.csv"
, "format": "edge_list"
, "vertex_id_type": "string"
, "vertex_labels": false
, "edge_label": false
, "vertex_props":[
{"name":"name", "type":"string"}
]
, "edge_props":[
{"name":"neighborhood", "type":"integer"}
, {"name":"fusion", "type":"integer"}
, {"name":"cooccurence", "type":"integer"}
, {"name":"coexpression", "type":"integer"}
, {"name":"experimental", "type":"integer"}
, {"name":"database", "type":"integer"}
, {"name":"textmining", "type":"integer"}
, {"name":"combined_score", "type":"integer"}
]
, "separator": ","
}
ちなみに前回と全く同じです(笑)
※今回これを用いるとタンパク質、薬剤のノードの情報の識別は出来ませんが、薬剤の方はID番号で表示されるので識別可能ということで考慮していません!
さて、読み込んでみます。
pgx > G = session.readGraphWithProperties("置いてあるディレクトリのパス/drug_ppi_edgelist.json")
PgxGraph[name=drug_ppi_edgelist,N=24493,E=11365453,created=1509003440476]
グラフ構造での読み込みに成功しました!良かった、、、
次回は入れたグラフ構造のデータ検索を簡単にしてみようと思います!それでは!