概要
こちらで話されていることを試してみました。
簡単に言うと、「Paired-Endのシーケンスデータで、クオリティーが低くく、
マージが上手く行かない際は、片方だけでTaxonomyしてみようぜ(非推奨だけど)」
ということです。
最近、マージが上手くいかないデータを解析した時にこのやり取りを参考にして、
「こういう発想があったのか」と思ったので試してみました。
こちらのデータを使用させて頂きました。
https://www.ncbi.nlm.nih.gov/sra/SRX18463592[accn]
0.準備
# sra-toolsインストールしておく
brew install sratoolkit
# オリジナルのデータを保存しておく場所
mkdir data
cd data/
# SRAを取ってきて、fastqにするまでしてもらう。
fasterq-dump -p SRR22498300
ls
> SRR22498300_1.fastq SRR22498300_2.fastq
# 圧縮しておく
for file in `ls *.fastq`; do
pigz $file
done
1.まずはふつうに
mkdir paired
cd paired
mkdir raw-data
cd raw-data/
ln -s ../../data/SRR22498300_1.fastq.gz ./sample1_S1_L001_R1_001.fastq.gz
ln -s ../../data/SRR22498300_2.fastq.gz ./sample1_S1_L001_R2_001.fastq.gz
# import
cd ..
qiime tools import \
--type "SampleData[PairedEndSequencesWithQuality]" \
--input-path ./raw-data \
--input-format CasavaOneEightSingleLanePerSampleDirFmt \
--output-path demux-paired-end.qza
# 可視化
qiime demux summarize --i-data demux-paired-end.qza --o-visualization demux-paired-end.qzv
qiime tools view demux-paired-end.qzv
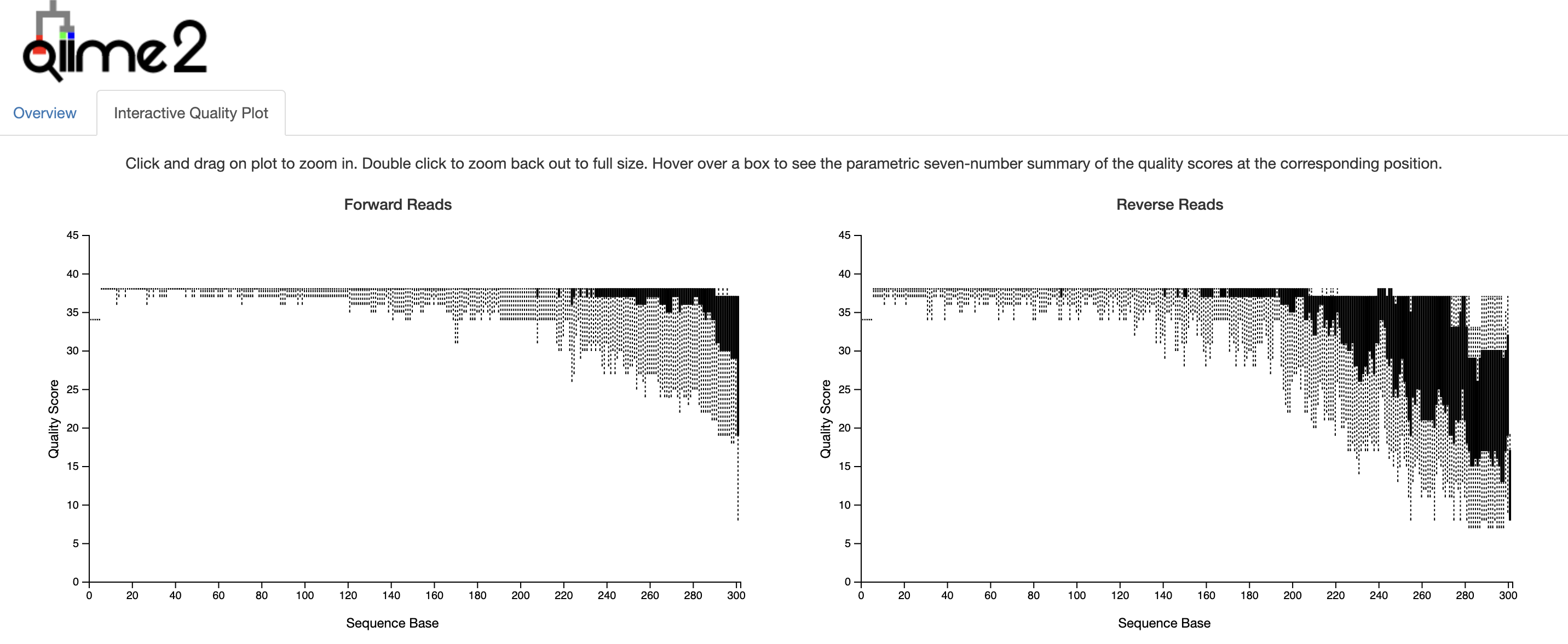
Forwardは300、Reverseは285あたりで切れば良さそうです。
qiime dada2 denoise-paired \
--i-demultiplexed-seqs ./demux-paired-end.qza \
--p-trunc-len-f 300 \
--p-trunc-len-r 285 \
--o-table table-dada2.qza \
--o-representative-sequences rep-seqs-dada2.qza \
--o-denoising-stats stats-dada2.qza \
--p-n-threads 16
# 可視化
qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv
qiime feature-table summarize \
--i-table table-dada2.qza \
--o-visualization table-dada2.qzv
qiime feature-table tabulate-seqs \
--i-data rep-seqs-dada2.qza \
--o-visualization rep-seqs-dada2.qzv
# 代表配列を見てみる
qiime tools view rep-seqs-dada2.qzv

平均453bpで、310配列配列取れているようです。
2. エラーを発生
mkdir -p error/raw-data
pigz -d data/SRR22498300_2.fastq.gz -c > data/SRR22498300_2.fastq
Reverseをだんだんと悪くする。
from Bio import SeqIO
import random
input_fastq = "./data/SRR22498300_2.fastq"
output_fastq = "./error/raw-data/sample1_S1_L001_R2_001.fastq"
records = SeqIO.parse(input_fastq, 'fastq')
new_records = []
# https://biotech-lab.org/articles/3133#SeqRecord
for record in records:
quality = record.letter_annotations['phred_quality']
changed = []
# ここらへんの数字は適当
top = 32
bottom = 20
for i, q in enumerate(quality):
if i % 20 == 0:
if top > 15:
top += -1
if bottom > 3:
bottom += -2
q = random.randint(bottom, top)
changed.append(q)
record.letter_annotations = {'phred_quality': changed}
new_records.append(record)
SeqIO.write(new_records, output_fastq, "fastq")
Forwardは3'側だけいじる
from Bio import SeqIO
import random
forward_input = "./data/SRR22498300_1.fastq"
forward_output = "./error/raw-data/sample1_S1_L001_R1_001.fastq"
records = SeqIO.parse(forward_input, 'fastq')
new_records = []
# https://biotech-lab.org/articles/3133#SeqRecord
for record in records:
quality = record.letter_annotations['phred_quality']
# 270辺りから15~35でランダム発生させる。
changed = []
for i, q in enumerate(quality):
if i > 270:
q = random.randint(15, 35)
changed.append(q)
record.letter_annotations = {'phred_quality': changed}
new_records.append(record)
SeqIO.write(new_records, forward_output, "fastq")
error/raw-data/ 下に、sample1_S1_L001_R2_001.fastqとsample1_S1_L001_R1_001.fastqが生成されるので、準備
cd error/raw-data/
# 圧縮
pigz sample1_S1_L001_R2_001.fastq
pigz sample1_S1_L001_R1_001.fastq
まずは普通にPaired-Endとしてimportしてみます。
# import
cd ..
qiime tools import \
--type "SampleData[PairedEndSequencesWithQuality]" \
--input-path ./raw-data \
--input-format CasavaOneEightSingleLanePerSampleDirFmt \
--output-path demux-paired-end.qza
# 可視化
qiime demux summarize --i-data demux-paired-end.qza --o-visualization demux-paired-end.qzv
qiime tools view demux-paired-end.qzv

ランダム発生の仕方が雑なので不自然すぎますが、いい感じに使いづらいデータにすることはできました。
Forwardは271、Reverseは20で切ってみます。
qiime dada2 denoise-paired \
--i-demultiplexed-seqs ./demux-paired-end.qza \
--p-trunc-len-f 271 \
--p-trunc-len-r 20 \
--o-table table-dada2.qza \
--o-representative-sequences rep-seqs-dada2.qza \
--o-denoising-stats stats-dada2.qza \
--p-n-threads 16
# 可視化
qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv
qiime feature-table summarize \
--i-table table-dada2.qza \
--o-visualization table-dada2.qzv
qiime feature-table tabulate-seqs \
--i-data rep-seqs-dada2.qza \
--o-visualization rep-seqs-dada2.qzv
# 代表配列を見てみる
qiime tools view rep-seqs-dada2.qzv
270bpで、1つしか出なかったようです。
(mergeが1つもできず、エラー発生することを想定したいたのですが...。)

では、本題のSingle-Endとして扱ってみます。
# 避難させる
mkdir error-paired
mv *.qza *.qzv error-paired/
mv raw-data/sample1_S1_L001_R2_001.fastq.gz error-paired/
一旦、指定可能なタイプを表示します。
qiime tools import --show-importable-types
Bowtie2Index
DeblurStats
DistanceMatrix
EMPPairedEndSequences
EMPSingleEndSequences
ErrorCorrectionDetails
FeatureData[AlignedProteinSequence]
FeatureData[AlignedRNASequence]
FeatureData[AlignedSequence]
FeatureData[BLAST6]
FeatureData[Differential]
FeatureData[Importance]
FeatureData[PairedEndRNASequence]
FeatureData[PairedEndSequence]
FeatureData[ProteinSequence]
FeatureData[RNASequence]
FeatureData[Sequence]
FeatureData[Taxonomy]
FeatureTable[Balance]
FeatureTable[Composition]
FeatureTable[Design]
FeatureTable[Frequency]
FeatureTable[PercentileNormalized]
FeatureTable[PresenceAbsence]
FeatureTable[RelativeFrequency]
Hierarchy
MultiplexedPairedEndBarcodeInSequence
MultiplexedSingleEndBarcodeInSequence
PCoAResults
Phylogeny[Rooted]
Phylogeny[Unrooted]
Placements
ProcrustesStatistics
QualityFilterStats
RawSequences
SampleData[AlphaDiversity]
SampleData[ArtificialGrouping]
SampleData[BooleanSeries]
SampleData[ClassifierPredictions]
SampleData[DADA2Stats]
SampleData[FirstDifferences]
SampleData[JoinedSequencesWithQuality]
SampleData[PairedEndSequencesWithQuality]
SampleData[Probabilities]
SampleData[RegressorPredictions]
SampleData[SequencesWithQuality]
SampleData[Sequences]
SampleData[TrueTargets]
SampleEstimator[Classifier]
SampleEstimator[Regressor]
SeppReferenceDatabase
TaxonomicClassifier
UchimeStats
Paired-Endで、SampleData[PairedEndSequencesWithQuality]としてimportしたので、
今回はSampleData[SequencesWithQuality]として扱います。
qiime tools import \
--type "SampleData[SequencesWithQuality]" \
--input-path ./raw-data \
--input-format CasavaOneEightSingleLanePerSampleDirFmt \
--output-path demux-single-end.qza
前回と同じく、271で切ってみます。
qiime dada2 denoise-single \
--i-demultiplexed-seqs ./demux-single-end.qza \
--p-trunc-len 271 \
--o-table table-dada2.qza \
--o-representative-sequences rep-seqs-dada2.qza \
--o-denoising-stats stats-dada2.qza \
--p-n-threads 16
# 可視化
qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv
qiime feature-table summarize \
--i-table table-dada2.qza \
--o-visualization table-dada2.qzv
qiime feature-table tabulate-seqs \
--i-data rep-seqs-dada2.qza \
--o-visualization rep-seqs-dada2.qzv
# 代表配列を見てみる
qiime tools view rep-seqs-dada2.qzv
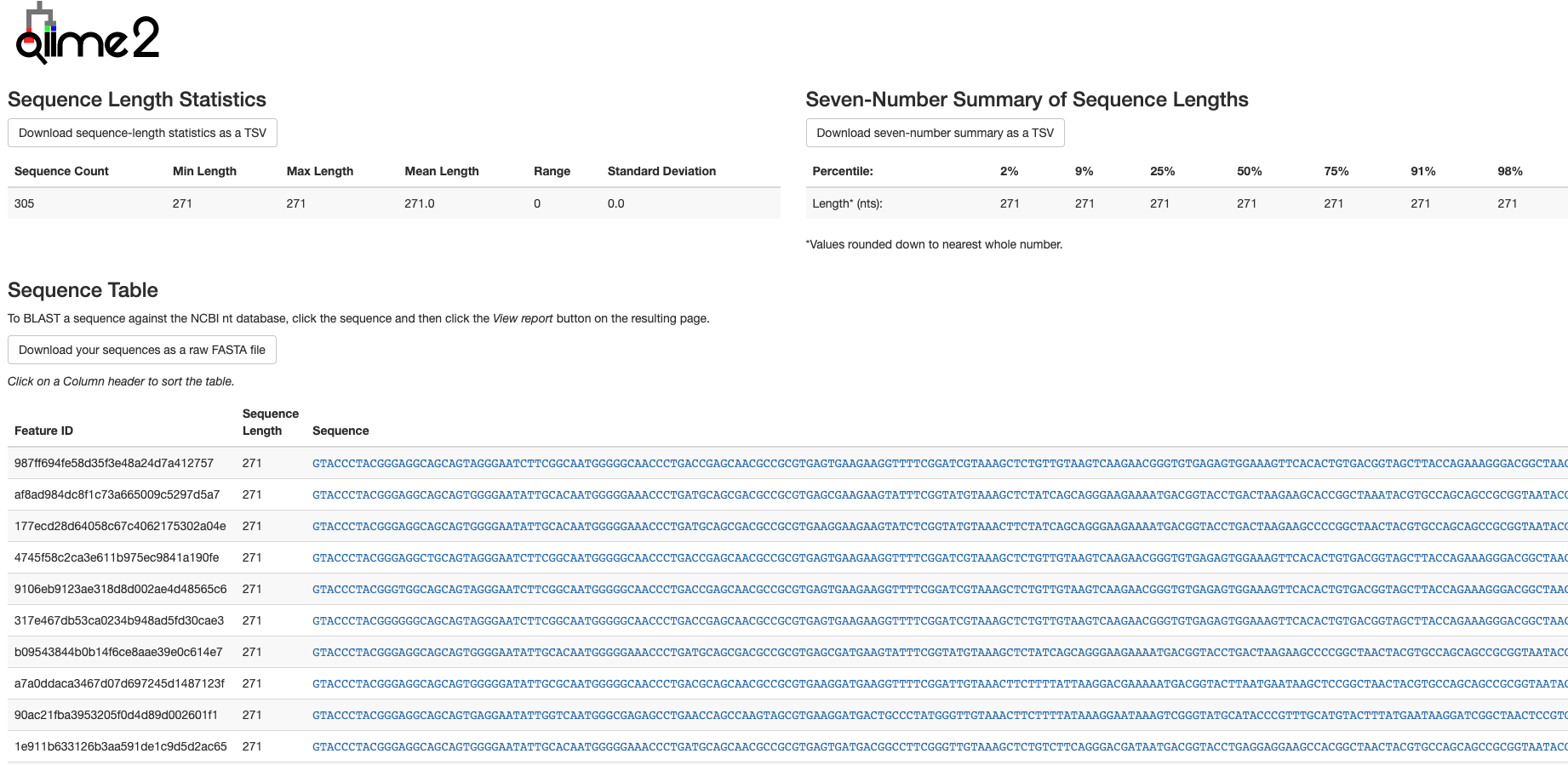
271bpの305配列取得することができました。
まとめ
今回はキレイな配列データの「クオリティのみ」をいじったので、そりゃあ上手くいくわという感じですが、
mergeする作業によって代表配列が作れない場合の応急処置としてのTipsでした。
もしこのようなデータにふれる機会があれば、実際うまくいくのか試してみたいと思います。
参考