- メタゲノム解析でいけてる手法としてWGCNA(Weighted Gene Correlation Network Analysis)によるネットワーク解析がある
- 海洋微生物学だとDeLongのチームがよく使っている 例: https://www.nature.com/articles/s41564-017-0008-3
- 元々はマイクロアレイデータとかを念頭として作られたが、最近メタゲノム分野への応用が増えてきてる印象
論文
- R実装について: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-559
- 式はこの論文から引用: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000117
- 理論の方の大元の論文はアクセスできなかった

TLDR
- 遺伝子発現のデータやメタゲノムデータをクラスタリングする手法の一つ(ネットワーク解析の手法としても使える)
- 重み付けした相関ネットワークに基づきクラスタリング
- 重みのパラメータ
soft thresholding powerの設定が肝 - サンプル数が多い時はかなり強力な手法っぽい
メタゲノムにおける相関ネットワーク解析とは?
- 相関する遺伝子やOTUに線を引き、ネットワークとして可視化する手法
- 先日同僚が出したbioRxivにいい感じの例があったので引用する
- Hiraoka, Satoshi, et al. "Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments." bioRxiv (2019): 729517. https://www.biorxiv.org/content/10.1101/729517v1
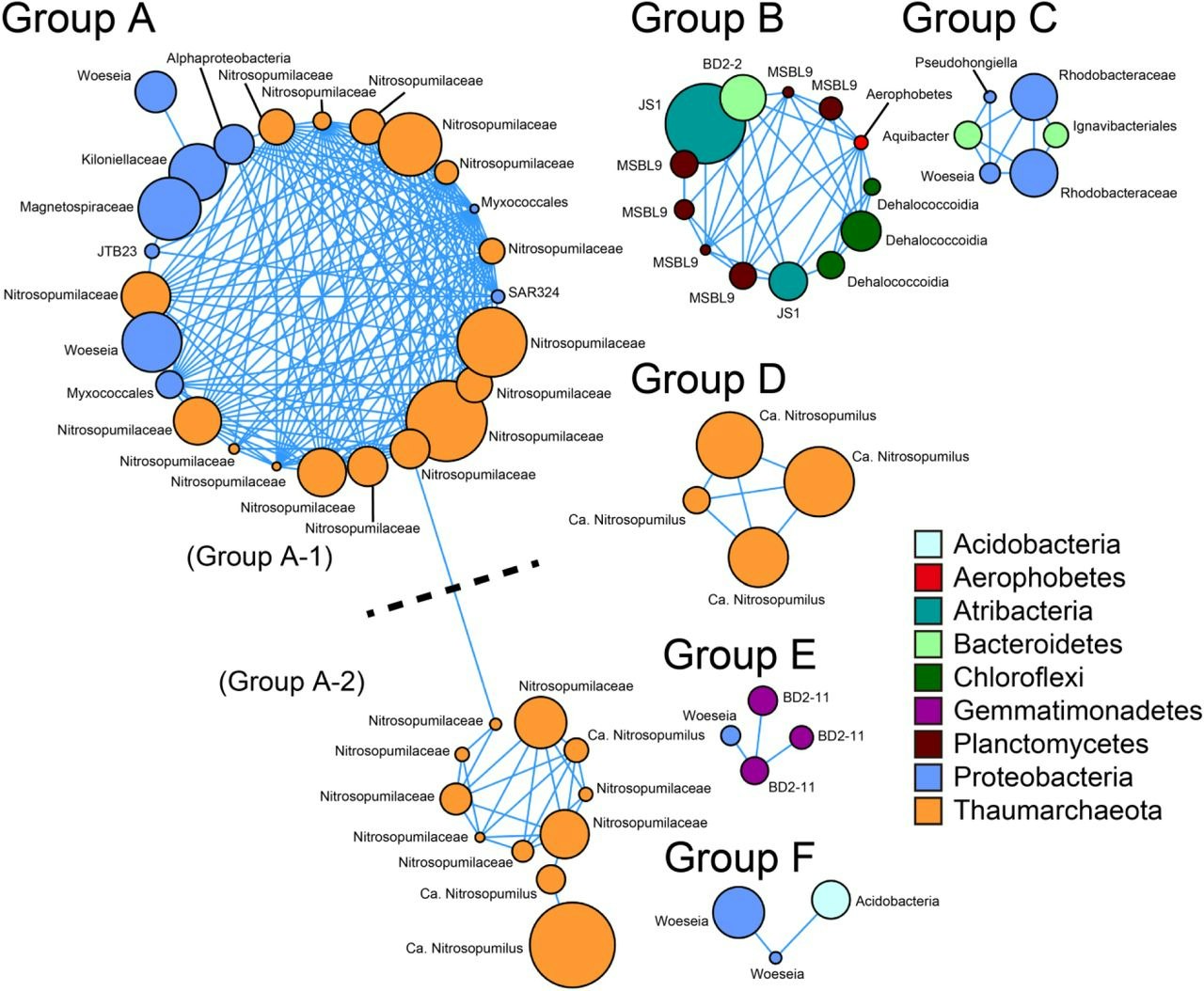
- 上の例ではメタ16SのOTUを使って、OTU間の相関係数 > 0.8の場合にOTU間に線を引いている
- 図のように独立したネットワークに名前を付けるとクラスタリングのような感じで使える
上記手法の問題点
- 相関係数 > 0.8 で切って[0, 1]の関係に直しているので、データのロスが大きい
- うまく独立したネットワークができない場合相関係数の閾値とかを微調整したりしょっぱい作業が必要
- クラスタリング部分が人間の直感に基づくところがあるのでもうちょい数学的にきちんと処理したい
- 結果クラスタリングが目的なら、ネットワークを図示する意味はあるのか?みたいな気もする(ネットワークの可視化はだいたいめちゃくちゃめんどくさい)
-> この辺りを解決するのがWGCNA
WGCNA
ネットワーク作成
-
適当な閾値を決めて相関係数を 1 or 0 のデータにするのではなく、WGCNAでは重み(コネクションの太さ)を考慮
-
ネットワークの重みには相関係数を直に使うのではなく soft thresholding power(β)を使い、(相関係数^β) で計算(β > 1)。生データは大体ノイジーかつサンプル数が十分でない状況にも対応できるよう、弱い相関は無視し強い相関を増幅するためにこうしているとのこと。参考
-
ネットワークの接続性(connectivity)と、スケールフリー性を考慮してβを設定する(この二つはtrade-off関係)。
-
Connectivity
-
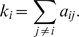
-
ある遺伝子に対する全ての遺伝子の相関係数の総和 (a_ijが相関係数)
-
スケールフリー性
-
「現実世界のネットワークが持つ第1の性質は「スケールフリー性」(次数分布のべき乗則)である。これは、一部の頂点が他のたくさんの頂点と辺で繋がっており、大きな次数を持っている一方で、その他の大部分はわずかな頂点としか繋がっておらず、次数は小さいという性質である。次数の大きな頂点は「ハブ」とも呼ばれる。...数学的には、スケールフリー性は頂点が次数 k を持つ確率 p(k) の確率分布が p(k) ∝ k-γ のべき乗則になる」wikipedia
-
(あまりちゃんとは理解していないが)「スケールフリー性」は「少数のhubが多数とつながるネットワーク構造」のこと。βをでかくすると強い相関しか残らなくなっていくので、この構造が強くなっていく。しかし極端に強い相関しか残らないとConnectivityが下がる(データを捨てている)。
-
WGCNAのチュートリアルでは
Scale independenceという言い方をしている -
Maximum adjacency ratio (MAR)
-
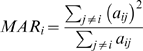
-
めっちゃ強い関係(a_ij = 1)とめっちゃ弱い関係 (a_ij ≈ 0)だけのときは (a_ij)と(a_ij)^2に差が生まれないので1に近づく。また、相関係数に重み付けをせず1 or 0とすると実質MAR = 1となる。クラスタのトポロジーに関する指標であり、MARiが大きい遺伝子->少ない遺伝子と強固に結合する、MARiが小さい遺伝子->多くの遺伝子と弱めの結合をする、というような判断ができる
-
eigengene
-
各モジュールごとにPCAした第一主成分。重みづけした発現量をモジュールごとに平均したような値となり、「そのモジュールの代表的な発現パターン」を示す。モジュールのHubとなる遺伝子はeigengeneと類似した発現パターンを示す
クラスタリング
-
ざっくりいうと「遺伝子AとBが、多くの共通する遺伝子とネットワーク上で繋がっている時、この二つは同じクラスタに属する」という感じ
-
クラスタリングに使う距離行列としてTopological overlapで計算した遺伝子の距離を用いる(ある二つの遺伝子同士が何個の遺伝子をシェアするかを距離とする)。距離なので実際には(1 - Topological overlap)。
-
遺伝子のクラスタリング(module detection)は単純に階層的クラスタリング(hclust)してからいい感じのheightで切る。
-
Topological overlap
-
論文: https://academic.oup.com/bioinformatics/article/23/2/222/205537
-
重みづけなしのネットワークにおける「ノードi, jが共有するネットワークの数」を重みづけしたネットワークに拡張したもの
R packageに関して
- Rの
WGCNAパッケージではmodule同士の関係性や、何かしらの条件と有意に関連するmoduleのdetectができる(この辺りはマイクロアレイの解析を主眼に作られている) - チュートリアルはちゃんとしているが理論そこそこ理解してないと使うのむずい
- 理論さえわかっていればコピペでほぼほぼいける
- 色分けする前提なのか、検出されたクラスタのid的なやつが
colorsとして渡されるなど仕様には不満がそこそこある