マイクロサテライトやRAD-seq、MIG-seqで得られた多型データから集団間の遺伝子流動を調べたい際に、Fastsimcoal2やDadiによるモデルベースの集団動態推定をするのはなかなかハードルが高いです。
BayesAssは集団間の最近(過去数世代)の移住率(m)について、マルコフ連鎖モンテカルロ法を用いて事後確率を推定するプログラムで、比較的手軽に解析が行えます。
ここでは、Stacksから出力された多型データを例に、BayesAssでの解析方法を簡単に解説します。
間違えがあればコメント等でご指摘下さい!
環境
macOS 11.4
Stacks 2.54
GenoDive 3.05
BayesAss 3.0.4.2
インプットファイルの準備
Stacks内のプログラムの一つであるpopulationsはカタログ化された遺伝子座から、Popmapファイルで指定された個体ごとに一定の基準で遺伝子座およびSNPを出力してくれます。
Stacksの使い方については、以下をご参考ください。
populationsでのジェノタイプ出力の際に、Genepop形式のファイルを出力するオプション--genepopを付けます。
populations -P ./stacks -M ./popmap.txt -O ./populations --write-single-snp --genepop -R 0.6 --min-mac 2 --max-obs-het 0.75 -t 16
これで以下のファイルが出力されます。
populations.snps.genepop
このファイルをBayesAssで読み込める形式に変換します。
フォーマット変換にはいくつかの方法がありますが、MacユーザーであればGenoDiveで変換するのが一番楽です。
・GenoDiveで変換
・PGDSpiderで変換
GenoDiveでの変換はごく簡単です。
メニューバーから
[Data] → [Convert to Other Format...]
そして、BayesAss formatを選択してConvertします。
最後に変換されたデータセットをBayesAss形式で書き出します。
[File] → [Export] → [BayesAss...]
保存したファイルの拡張子は.txtに変えておきましょう。
BayesAssでの解析
BayesAssは以下のサイトから予めダウンロードしておきます。
MacユーザーはHomebrewを使ってインストールできます。
brew tap brannala/ba3
brew install ba3
コンパイル等は不要です。そのまま動きます。
パッケージには以下の2つのプログラムが含まれています。
BA3-OSX64-MSAT(ba3msat)
BA3-OSX64-SNP(ba3snp)
# カッコはHomebrewでインストールした場合
BA3-SNPは最大で30,000遺伝子座、1座あたり4対立遺伝子、20集団、2000個体の使用一塩基多型(SNP)データ、BA3-MSATは最大で500遺伝子座、1座あたり500対立遺伝子、100集団、5000個体のマイクロサテライトデータに対応しているようです。
基本的な解析の実行は以下のようにします。
ba3snp -v -t -g -i10000000 -b1000000 -n1000 input.txt
# -a --deltaA 対立遺伝子頻度の混合パラメータ値
# -b --burnin Burn-inとして捨てるMCMCの反復数
# -f --deltaF 近交係数の混合パラメータ値
# -g --genotypes 遺伝子型と移住祖先の出力
# -i --iterations MCMCの反復回数
# -m --deltaM 移住率の混合パラメータ値
# -n --sampling MCMCのサンプリング頻度
# -o --output 出力ファイル名
# -s --seed 乱数生成用のシード値
# -p --nolikelihood 尤度を1に固定して事前分布を生成
# -t --trace 収束をチェックするためのトレースファイルの出力
# -u --settings オプションとパラメータ設定を出力
# -v --verbose 冗長出力の使用
3種類の混合パラメータ調整は、それぞれ対立遺伝子頻度、近親交配係数、移住率の提案サイズを調整し、0に近い値では変化が小さく、1に近い値では変化が大きくなるようです。ただし、BA3では、0.35から0.45の間のMCMC採択率を目標とし、最終的な採択率がこの範囲内に収まるように自動調整してくれます(本当かなぁ・・・)。
logP(M): -159.75 logL(G): -8078.64 logL: -8238.39 % done: [0.10] % accepted: (0.59, 0.00, 0.76, 0.68, 0.48)
logP(M): -163.52 logL(G): -7890.20 logL: -8053.72 % done: (0.11) % accepted: (0.59, 0.00, 0.76, 0.68, 0.48)
logP(M): -170.80 logL(G): -7863.66 logL: -8034.46 % done: (0.12) % accepted: (0.59, 0.00, 0.76, 0.68, 0.48)
問題がなければこんな感じの出力がしばらく続きます。
logP(M)は、現在の移住率を条件として、個体間で移住者の祖先がどのように構成されているかの対数確率です。
logL(G)は、個体の移住祖先と現在の母集団の対立遺伝子頻度が与えられたときの遺伝子型データの対数尤度です。
logLは、これら2つの値を合計したものです。
doneは、全反復のうち完了したものの割合です。Buin-in中の場合は[角括弧]で割合が示されます。
acceptedにある値は、左から移住率(migration rates)、個体移住祖先(individual migrant ancestries)、対立遺伝子頻度(allele frequencies)、近親交配係数(inbreeding coefficients)、遺伝子型のミッシング(missing genotypes)の採択率を表しています。
このうち、1、3、4番目の採択率が最終的に20%〜60%の間にくるのが最適であるとされています。
採択率が低すぎても高すぎても推定がうまくできません。
混合パラメータのデフォルト値は0.10なので、採択率が高すぎる場合は-a、-f、-mオプションで初期値を高く、低すぎる場合は小さくして再度解析します。
上の例では3つの採択率がすべてが高すぎるので、混合パラメーターの初期値をあげて再度解析します。
./BA3-OSX64-SNP -v -t -g -i1000000 -b100000 -n10000 -m 0.3 -a 0.5 -f 0.3 populations.snps.txt
# -m --deltaM 移住率の混合パラメータ値
# -a --deltaA 対立遺伝子頻度の混合パラメータ値
# -f --deltaF 近交係数の混合パラメータ値
logP(M): -162.58 logL(G): -8151.51 logL: -8314.08 % done: (0.10) % accepted: (0.35, 0.00, 0.45, 0.39, 0.53)
logP(M): -161.68 logL(G): -7990.89 logL: -8152.57 % done: (0.20) % accepted: (0.36, 0.00, 0.46, 0.41, 0.50)
logP(M): -164.63 logL(G): -8018.73 logL: -8183.36 % done: (0.30) % accepted: (0.36, 0.00, 0.47, 0.41, 0.49)
かなり良くなりました。
収束の確認
解析がちゃんと収束していることをベイズ解析でお馴染みのTracerで確認します。
-tオプションをつけることで出力されるBA3trace.txtファイルをTracerで読み込み、対数確率(LogProb)が急激に増減することなく安定していることを確認します。
あくまで目安ですが、LogProbのESSが200を超えていればOKです。
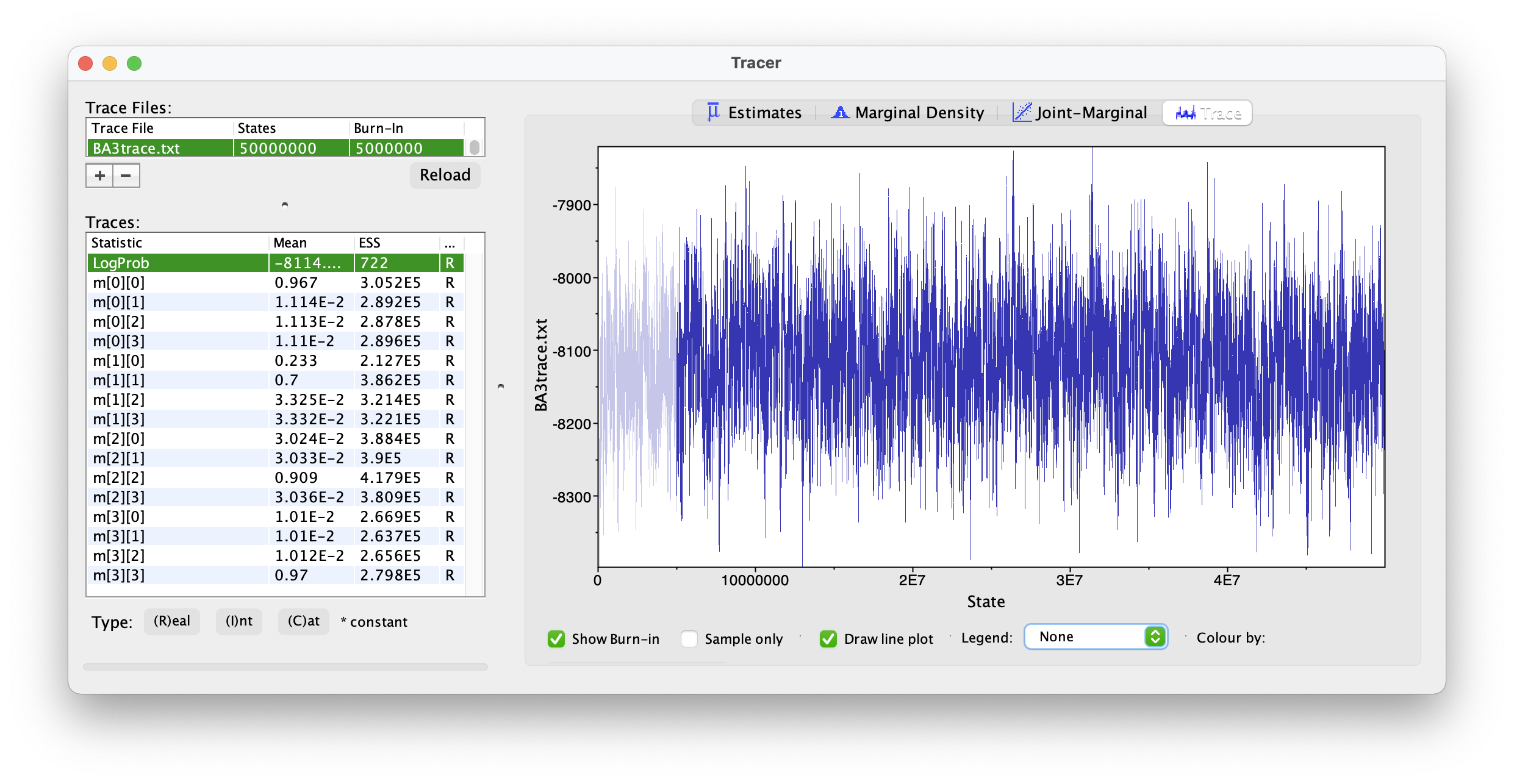
収束していない場合は、パラメーターの採択率が20%〜60%の間にあるかを確認し、ない場合は初期値を調整します。
また、MCMCの回数やサンプリングの頻度を増やして再解析します。
結果の解釈と注意点
アウトプットファイルの中身は以下のようになっています。
Input file: examples/3pop.txt
Individuals: 400 Populations: 3 Loci: 7
Locus:(Number of Alleles)
loc0:2 loc1:3 loc2:3 loc3:3 loc4:2 loc5:2 loc6:2
Population Index -> Population Label:
0->pop0 1->pop1 2->pop2
Migration Rates:
m[0][0]: 0.9718(0.0115) m[0][1]: 0.0130(0.0100) m[0][2]: 0.0152(0.0090)
m[1][0]: 0.0878(0.0141) m[1][1]: 0.7338(0.0396) m[1][2]: 0.1784(0.0407)
m[2][0]: 0.0870(0.0179) m[2][1]: 0.2047(0.0326) m[2][2]: 0.7083(0.0292)
Inbreeding Coefficients:
pop0 Fstat: 0.2552(0.0367)
pop1 Fstat: 0.0810(0.0588)
pop2 Fstat: 0.2698(0.0891)
Allele Frequencies:
pop0
loc0>>
2:0.806(0.029) 1:0.194(0.029)
loc1>>
3:0.309(0.033) 2:0.601(0.035) 1:0.089(0.020)
loc2>>
2:0.751(0.031) 3:0.170(0.027) 1:0.078(0.018)
loc3>>
2:0.384(0.035) 1:0.292(0.032) 3:0.324(0.032)
loc4>>
1:0.825(0.028) 2:0.175(0.028)
loc5>>
2:0.234(0.031) 1:0.766(0.031)
loc6>>
2:0.237(0.031) 1:0.763(0.031)
pop1
...
Migration Ratesのあとに書かれている部分が最も知りたい情報です。
m[i][j]は、集団iの個体のうち、集団jからの移住に由来する割合(1世代あたり)を表します。括弧の中は周辺事後分布の標準偏差。
その下には、各集団の近交係数、対立遺伝子頻度の平均事後推定値(と標準誤差)が出力されています。
注意!
ここで推定されたmは移住率であって移住個体数ではないことに注意が必要!
(集団サイズが小さい時は、移住率が高かった場合であっても移住個体数は少ないと考えられる)
結果の図示
BayesAssの結果はChord Diagramやヒートマップにしたり、地図上に矢印で示したりするのが一般的かと思います。
引用文献・サイト
- Wilson GA, Rannala B. 2003. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191 https://doi.org/10.1093/genetics/163.3.1177
- Mussmann SM, Douglas MR, Chafin TK, Douglas ME. 2019. BA3-SNPs: Contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol Evol. 10: 1808–1813 https://doi.org/10.1111/2041-210X.13252
- BayesAssのマニュアル:https://github.com/brannala/BA3/blob/master/doc/BA3Manual.pdf
Qiitaにも、参考になる解説があります。