HiSeqなどのイルミナシーケンサーで得られた全ゲノムリシーケンスデータからミトコンドリアや葉緑体といったオルガネラゲノムをアセンブルする方法について備忘録を兼ねて簡単に解説します。この手法はプライマーの開発や複数回のPCR・シーケンスが不要なため、コストパフォーマンスに優れており、最近では主流のオルガネラゲノム決定法となっています。
ここではスピーディなNOVOplastyと時間はかかるがより正確なGetOrganelleのふたつのアセンブラの使い方紹介します。
環境
NOVOPlasty 4.3.5
GetOrganelle 1.7.7.1
Python 3.12.2
MacOS 14.4.1
Apple M2 Max
※古いバージョンでの実行結果があります。
- 解説はMacOSの場合で進めていきますが、WindowsやLinuxでも動かせます。AppleシリコンのPCでも動きます。
- アセンブルにはある程度のPCスペックが要求されます(特にメモリが重要、32GBは欲しい)。
- 一部、Mac用のパッケージマネージャであるHomebrewを利用します。
- NOVOplastyにはperl、GetOrganelleにはPython3が必要です。
インプットデータの準備
動物のミトコンドリアゲノムの場合、データ量は2〜3GBぐらいあれば十分です(150PEなら各500万リードぐらい?)。データ量が多すぎる場合は、解析時間が無駄にかかるので、データの一部だけを抜き出します。
リードのクオリティコントロールは基本的に不要ですが、アダプターやインデックスが含まれている場合はfastpなどを用いて事前にトリミングしておきます。
薄ゲノムなど、既にデータ量が少ない場合はこの作業は不要です。
GetOrganelleのv1.6.2以降では、必要なリードデータを自動で推定してくれるので、インプットのデータ量が多いからといって解析時間が極端に多くなることはないようです。
ここではSeqkitを利用します。
インストールはHomebrewで簡単にできます。
brew install seqkit #Seqkitのインストール
seqkit head -n 5000000 ./Fish_rawdata_R1.fq.gz > Fish_5000000_R1.fq | gzip Fish_5000000_R1.fq
seqkit head -n 5000000 ./Fish_rawdata_R2.fq.gz > Fish_5000000_R2.fq | gzip Fish_5000000_R2.fq
#頭の500万リードを抽出し、再圧縮
これでインプットファイルの準備は完了です。
アセンブラの準備
データファイルの準備ができたら、アセンブラをダウンロードします。
Homebrewにはないので、GitHubから落とします。
git clone https://github.com/ndierckx/NOVOPlasty.git #NOVOplastyのダウンロード
#NOVOplastyはインストール作業不要
git clone https://github.com/Kinggerm/GetOrganelle.git #GetOrganelleのダウンロード
pip3 install ./GetOrganelle #GetOrganelleのインストール
#pipはPythonについてるパッケージ管理ツール
pipを使わないインストール方法については公式を参照。
GetOrganelleでは3つのプログラムSPAdes, Bowtie2, BLAST+が要求されるので、これらも事前にインストールしておきます。
brew install numpy scipy spades Bowtie2 blast
また、Pythonライブラリのnumpy, sympy, scipy, requestsも必要になりますので、まだ入っていない場合はインストールしておきます。
これでアセンブラの準備は完了です。
NOVOplastyの使い方
事前にConfigファイルを作成する必要があります(サンプルごとに必要)。
テンプレートがあるので、それを複製して使いまわします。
$ cat config.txt
Project:
-----------------------
Project name = Test
Type = mito #葉緑体ならchloro、植物ミトコンならmito_plant
Genome Range = 14000-20000
K-mer = 39 #アセンブルの成否はこの値で決まる。うまくいかない場合はリード長やデータ量によって増減させる
Max memory =
Extended log = 0
Save assembled reads = no
Seed Input = /path/to/seed_file/Seed.fasta #同種や近縁種のミトコンドリアDNAの一部(COI配列など)を指定
Reference sequence = /path/to/reference_file/reference.fasta #もし同種のミトゲノムが決まっているなら指定(なくてもOK)
Variance detection =
Chloroplast sequence = #Typeがmito_plantの時のオプション
Dataset 1:
-----------------------
Read Length = 151 #リード長
Insert size = 300 #大体のインサートサイズ
Platform = illumina #使ったシーケンサーのメーカー
Single/Paired = PE
Combined reads =
Forward reads = /path/to/reads/reads_1.fastq #インプットデータ(リード1)
Reverse reads = /path/to/reads/reads_2.fastq #インプットデータ(リード2)
Heteroplasmy: #最初はいじらなくていい
-----------------------
MAF =
HP exclude list =
PCR-free =
Optional: #最初はいじらなくていい
-----------------------
Insert size auto = yes
Insert Range = 1.9
Insert Range strict = 1.3
Use Quality Scores = no
初期状態では、Reference sequenceなどに例として/path/to/reference_file/reference.fasta (optional)のような文字列が入っていますが、不要な部分についてはエラーの原因となるので消しておきます。
Configファイルができたらあとはperl NOVOPlastyx.x.pl -c config.txtでRunするだけ。
$ perl NOVOPlasty3.6.pl -c config.txt #configファイルを指定するだけ
-----------------------------------------------
NOVOPlasty: The Organelle Assembler
Version 3.6
Author: Nicolas Dierckxsens, (c) 2015-2019
-----------------------------------------------
Input parameters from the configuration file: *** Verify if everything is correct ***
Project:
----------------------
Project name = test-fish
Type = mito
Genome range = 14000-19000
K-mer = 21
Max memory =
Extended log = 0
Save assembled reads = no
Seed Input = ~/seed-seq/AB000000.1_test-fish_COI.fasta
Reference sequence =
Variance detection =
Chloroplast sequence =
Dataset 1:
----------------------
Read Length = 150
Insert size = 300
Platform = illumina
Single/Paired = PE
Combined reads =
Forward reads = ~/rawdata/test-fish_R1.fastq.gz
Reverse reads = ~/rawdata/test-fish_R2.fastq.gz
Heteroplasmy:
-----------------------
Heteroplasmy =
HP exclude list =
PCR-free =
Optional:
----------------------
Insert size auto = yes
Insert range = 0.9
Insert range strict = 1.3
Use Quality Scores =
Subsampled fraction: 100.00 %
Retrieve Seed...
Initial read retrieved successfully:
GCCGGTCTTGTAATCCGGAGATCGAAGGTTAAAATCCTTCCTAGTGCCAGAATGTATCTAGTATTTGGTGCTTGAGCCGGAATAGTAGGGACCGCATTAAGCCTCTTAATCCGAGCTGAACTTAGCCAACCAGGAGCACTTCTTGGTGATG
#捏造配列です
Start Assembly...
-----------------Assembly 1 finished successfully: The genome has been circularized-----------------
Contig 1 : 16528 bp
Total contigs : 1 #Contigの数が1で環状になれば成功です
Largest contig : 16528 bp
Smallest contig : 16528 bp
Average insert size : 258 bp
-----------------------------------------Input data metrics-----------------------------------------
Total reads : 3715536
Aligned reads : 2746
Assembled reads : 2586
Organelle genome % : 0.07 %
Average organelle coverage : 25 #平均デプス
-----------------------------------------------------------------------------------------------------
環状になったミトゲノムがfasta形式で出力されます。
早ければ5分ほどで終わります。データ量が多い場合や、K-merの値が適当でない場合は時間がかかります。
うまくアセンブルされない場合は、インプットデータのQCやデータ量の増減、K-mer値の増減、シード配列の変更を検討します。
NOVOplastyのいいところ
- 超早い
- シンプル
NOVOplastyの問題点
- K-merを変えて試行錯誤するのが面倒くさい
- アセンブルがうまくいかない個体がでてくる
そこでより正確で多機能なGetOrganelleの出番です。
GetOrganelleの使い方
バージョン1.7.0からリファレンス配列のデータベースが独立したので、まずは対象分類群のDBを読み込むところからはじめます。
get_organelle_config.py -a animal_mt
#6つのDBが用意されているのでこの中から選ぶ
#embplant_pt, embplant_mt, embplant_nr, fungus_mt, animal_mt, other_pt
#適当なDBがない場合は自分で用意したファイルを-sおよび--geneオプションで指定できる
NOVOplastyのようなConfigファイルはありません。
$ get_organelle_from_reads.py -1 ~/rawdata/test-fish_R1.fastq.gz -2 ~/rawdata/test-fish_R2.fastq.gz -o test-fish -R 10 -k 21,45,65,85,105 -F animal_mt
-1 #リード1のインプットファイル、-2でリード2のインプットファイルを指定
-o #出力ファイル名
-R #Extendingのラウンド数。何をしているのかよくわからないが10〜15ぐらいがいい
-k #K-merの値を指定。リード長より大きくしないこと
-F #オルガネラゲノムの種類を指定。動物だったらanimal_mt、有胚植物の葉緑体だったらembplant_pt。公式参照
-s #シード配列を指定。-Fで既存のデータベースを参照している場合は不要
-genes #遺伝子配列を指定。-Fで既存のデータベースを参照している場合は不要
GetOrganelle v1.7.1a
get_organelle_from_reads.py assembles organelle genomes from genome skimming data.
Find updates in https://github.com/Kinggerm/GetOrganelle and see README.md for more information.
Python 3.6.8 (v3.6.8:3c6b436a57, Dec 24 2018, 02:04:31) [GCC 4.2.1 Compatible Apple LLVM 6.0 (clang-600.0.57)]
PYTHON LIBS: GetOrganelleLib 1.7.1a; numpy 1.16.4; sympy 1.5; scipy 1.4.1; psutil 5.7.2
DEPENDENCIES: Bowtie2 2.4.1; SPAdes genome assembler 3.14.1; Blast 2.10.1
SEED DB: animal_mt 0.0.0
LABEL DB: animal_mt 0.0.0
WORKING DIR: /Applications/GetOrganelle1.7.1
/Library/Frameworks/Python.framework/Versions/3.6/bin/get_organelle_from_reads.py -1 /Users/user/rawdata/test-fish_R1.fastq.gz -2 /Users/user/rawdata/test-fish_R2.fastq.gz -o test-fish -R 10 -k 21,45,65,85,105 -F animal_mt --memory-unlimited -t 16
2020-08-30 03:48:07,806 - INFO: Pre-reading fastq ...
2020-08-30 03:48:07,806 - INFO: Estimating reads to use ... (to use all reads, set '--reduce-reads-for-coverage inf')
2020-08-30 03:48:07,873 - INFO: Tasting 100000+100000 reads ...
2020-08-30 03:48:12,710 - INFO: Estimating reads to use finished.
2020-08-30 03:48:12,710 - INFO: Unzipping reads file: /Users/user/rawdata/test-fish_R1.fastq.gz (1178949358 bytes)
2020-08-30 03:48:14,715 - INFO: Unzipping reads file: /Users/user/rawdata/test-fish_R2.fastq.gz (1316414966 bytes)
2020-08-30 03:48:16,987 - INFO: Counting read qualities ...
2020-08-30 03:48:17,128 - INFO: Identified quality encoding format = Illumina 1.8+
2020-08-30 03:48:17,184 - INFO: Mean error rate = 0.0076
2020-08-30 03:48:17,184 - INFO: Counting read lengths ...
2020-08-30 03:48:21,569 - INFO: Mean = 151.0 bp, maximum = 151 bp.
2020-08-30 03:48:21,569 - INFO: Reads used = 1913222+1913222
2020-08-30 03:48:21,569 - INFO: Pre-reading fastq finished.
2020-08-30 03:48:21,569 - INFO: Making seed reads ...
2020-08-30 03:48:21,569 - INFO: Seed bowtie2 index existed!
2020-08-30 03:48:21,569 - INFO: Mapping reads to seed bowtie2 index ...
2020-08-30 03:48:49,676 - INFO: Mapping finished.
2020-08-30 03:48:49,677 - INFO: Seed reads made: test-fish/seed/animal_mt.initial.fq (117853 bytes)
2020-08-30 03:48:49,677 - INFO: Making seed reads finished.
2020-08-30 03:48:49,677 - INFO: Checking seed reads and parameters ...
2020-08-30 03:48:49,677 - INFO: The automatically-estimated parameter(s) do not ensure the best choice(s).
2020-08-30 03:48:49,677 - INFO: If the result graph is not a circular organelle genome,
2020-08-30 03:48:49,677 - INFO: you could adjust the value(s) of '-w'/'-R' for another new run.
2020-08-30 03:48:51,267 - INFO: Pre-assembling mapped reads ...
2020-08-30 03:48:52,453 - INFO: Pre-assembling mapped reads finished.
2020-08-30 03:48:52,453 - INFO: Estimated animal_mt-hitting base-coverage = 7.55
2020-08-30 03:48:52,567 - WARNING: Guessing that you are using too few data for assembling animal_mt!
2020-08-30 03:48:52,567 - WARNING: GetOrganelle is still trying ...
2020-08-30 03:48:52,568 - INFO: Estimated word size(s): 41
2020-08-30 03:48:52,568 - INFO: Setting '-w 41'
2020-08-30 03:48:52,568 - INFO: Setting '--max-extending-len inf'
2020-08-30 03:48:52,636 - INFO: Checking seed reads and parameters finished.
2020-08-30 03:48:52,636 - INFO: Making read index ...
2020-08-30 03:49:10,562 - INFO: Mem 1.651 G, 3507348 candidates in all 3826444 reads
2020-08-30 03:49:10,568 - INFO: Pre-grouping reads ...
2020-08-30 03:49:10,568 - INFO: Setting '--pre-w 41'
2020-08-30 03:49:10,873 - INFO: Mem 1.642 G, 289469/289469 used/duplicated
2020-08-30 03:49:50,375 - INFO: Mem 9.111 G, 52636 groups made.
2020-08-30 03:49:50,640 - INFO: Making read index finished.
2020-08-30 03:49:50,641 - INFO: Extending ...
2020-08-30 03:49:50,641 - INFO: Adding initial words ...
2020-08-30 03:49:50,658 - INFO: AW 27786
2020-08-30 03:50:43,196 - INFO: Round 1: 3507348/3507348 AI 743609 AW 42671528 Mem 8.176
2020-08-30 03:51:16,271 - INFO: Round 2: 3507348/3507348 AI 913396 AW 54972008 Mem 9.431
2020-08-30 03:51:43,856 - INFO: Round 3: 3507348/3507348 AI 950631 AW 57923292 Mem 9.578
2020-08-30 03:52:10,622 - INFO: Round 4: 3507348/3507348 AI 960259 AW 58687064 Mem 9.598
2020-08-30 03:52:37,149 - INFO: Round 5: 3507348/3507348 AI 963236 AW 58925054 Mem 9.603
2020-08-30 03:53:03,507 - INFO: Round 6: 3507348/3507348 AI 964298 AW 59008912 Mem 9.605
2020-08-30 03:53:29,792 - INFO: Round 7: 3507348/3507348 AI 964713 AW 59039266 Mem 9.606
2020-08-30 03:53:56,136 - INFO: Round 8: 3507348/3507348 AI 964831 AW 59047422 Mem 9.606
2020-08-30 03:54:22,480 - INFO: Round 9: 3507348/3507348 AI 964882 AW 59051312 Mem 9.606
2020-08-30 03:54:48,762 - INFO: Round 10: 3507348/3507348 AI 964903 AW 59052654 Mem 9.606
2020-08-30 03:54:48,763 - INFO: Hit the round limit 10 and terminated ...
2020-08-30 03:55:13,546 - INFO: Extending finished.
2020-08-30 03:55:13,577 - INFO: Separating filtered fastq file ...
2020-08-30 03:55:16,495 - INFO: Setting '-k 21,45,65,85,105'
2020-08-30 03:55:16,495 - INFO: Assembling using SPAdes ...
2020-08-30 03:55:16,518 - INFO: spades.py -t 16 -1 test-fish/filtered_1_paired.fq -2 test-fish/filtered_2_paired.fq --s1 test-fish/filtered_1_unpaired.fq --s2 test-fish/filtered_2_unpaired.fq -k 21,45,65,85,105 -o test-fish/filtered_spades
2020-08-30 04:43:56,532 - INFO: Insert size = 264.745, deviation = 85.871, left quantile = 157, right quantile = 379
2020-08-30 04:43:56,532 - INFO: Assembling finished.
2020-08-30 04:44:18,740 - INFO: Slimming test-fish/filtered_spades/K105/assembly_graph.fastg finished!
2020-08-30 04:44:18,740 - INFO: Slimming assembly graphs finished.
2020-08-30 04:44:18,740 - INFO: Extracting animal_mt from the assemblies ...
2020-08-30 04:44:18,741 - INFO: Disentangling test-fish/filtered_spades/K105/assembly_graph.fastg.extend-animal_mt.fastg as a circular genome ...
2020-08-30 04:44:18,775 - INFO: Average animal_mt kmer-coverage = 5.2
2020-08-30 04:44:18,775 - INFO: Average animal_mt base-coverage = 16.7
2020-08-30 04:44:18,775 - INFO: Writing output ...
2020-08-30 04:44:18,782 - INFO: Writing PATH1 of complete animal_mt to test-fish/animal_mt.K105.complete.graph1.1.path_sequence.fasta
2020-08-30 04:44:18,782 - INFO: Writing GRAPH to test-fish/animal_mt.K105.complete.graph1.selected_graph.gfa
2020-08-30 04:44:18,789 - INFO: Result status of animal_mt: circular genome
2020-08-30 04:44:18,808 - INFO: Please visualize test-fish/filtered_K105.assembly_graph.fastg.extend-animal_mt.fastg using Bandage to confirm the final result.
2020-08-30 04:44:18,808 - INFO: Writing output finished.
2020-08-30 04:44:18,809 - INFO: Extracting animal_mt from the assemblies finished.
Total cost 3371.82 s
Thank you!
Fasta形式のミトゲノム配列やアノテーション結果やグラフが出力されます。
最適パラメータの推定やマッピングなど色々してくれます。
オルガネラゲノムの構造の図示もできます(FastgファイルをBandageで開く)。
Thank you!
GetOrganelleのいいところ
- 正確
- パラメータ変えたり試行錯誤しなくていい
GetOrganelleの問題点
- すごい時間がかかる
NOVOplastyでもGetOrganelleでもミトゲノムが決まらない場合が稀にあります。その場合も調節領域だけ決まらない、みたいなパターンがほとんどです。
コンタミ等によって複数のオルガネラゲノムがアセンブルされる場合があります(エラーメッセージが出ます)。コンタミしたのが別種の場合、得られたコンティグをBLASTにかけることで、何が混ざったのか明らかにできるかもしれません。
こういった場合はマッピングツールを使ってリファレンス配列にリードを貼り付けることでミトゲノムを決定するのがおすすめです。
ゲノムアノテーション
最後は遺伝子領域を同定してフィニッシュです。
アノテーションはウェブベースで簡単にできます。
- MitoAnnotator(魚類;図示までしてくれる)
- MITOS
- MFannot
- DOGMA(提供終了)
Fastaファイルをポン入れして、はい簡単。
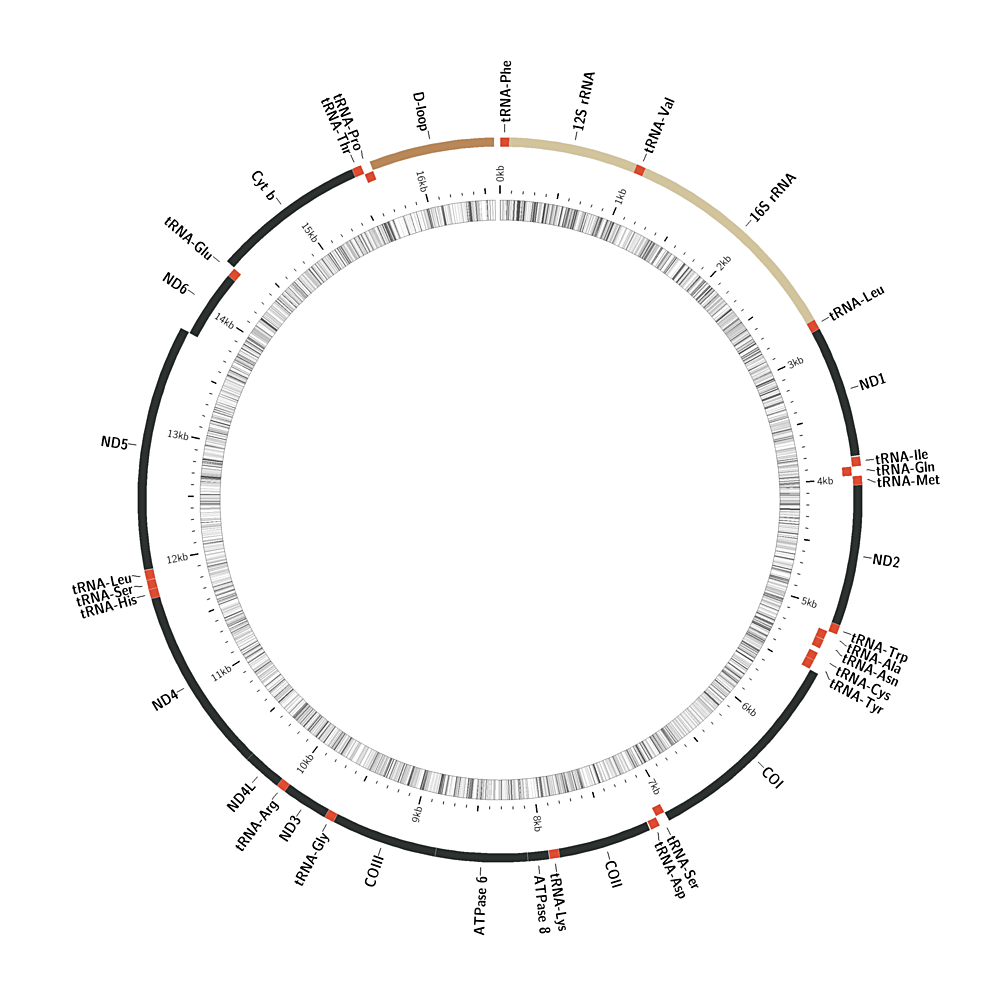
上手にできました!
文献
- Dierckxsens N, Mardulyn P, Smits G (2016) NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45(4): e18 https://doi.org/10.1093/nar/gkw955
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ (2018) GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv, 256479. http://doi.org/10.1101/256479