原文
ケモインフォマティクスにおける深層学習: 薬品分子の水溶性の予測 (Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules)
Alessandro Lusci, Gianluca Pollastri, and Pierre Baldi (2013)
1. 要約/背景
- DAG(有向非循環グラフ)-RNNを用いて、水の溶解度を予測した。
- 過去にも、浅層NNやSVMを使った予測アルゴリズムは考案されていた。利用データの量と質が広がるにつれて、それを深層学習器に落とし込んだ点が進歩である。
- そもそも論として、分子構造がNNやボルツマンマシンのそれと親和的であり、モデル適用のインスピレーションは高い。
2. 骨子の理論
(1)データの準備
入力データであるが、分子構造をUG(Undirected Graph, 無向グラフ)に落とし込んだものが使われている。(従来の方法では、融点、水ーオクタノール分配係数、原子価といった尤もらしいデータが入力データの特徴量として使われていた)
有機化学の見地では、「原子軌道法」という分子構造の記述に対応している。電子雲などの電子軌道の表現ができないため、実際の分子構造の再現性はやや劣る。とはいえ、非専門家にも理解しやすく、再現性も一定の水準で担保されるので、モデル起用の点では適している。ちなみに、分子薬学には構造活性相関という概念がある。本稿のモデル化に対するモチベーションも、分子構造と溶解度の間に相関性があるという考えがあってのことである。
$$\mbox{Acticity} = F(\mbox{structure}) = M(E(\mbox{structure}))$$
(2)分子モデリング
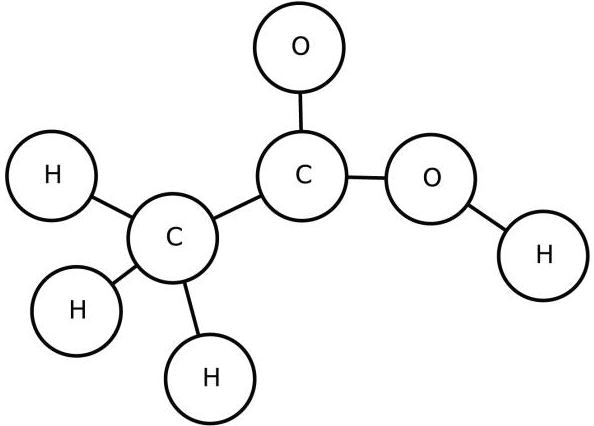
原子一つを一つのノードと見て、UGを描く。**環構造(芳香環など)は、1つの環を1つのノードを見る。**下記は、酢酸のUGである。
(3)DAG-RNN
構造の特徴学習からはじめるが、それはRNNを使う。DAGは入力値の特徴を見つけ出すための事前学習に使用する概念である。
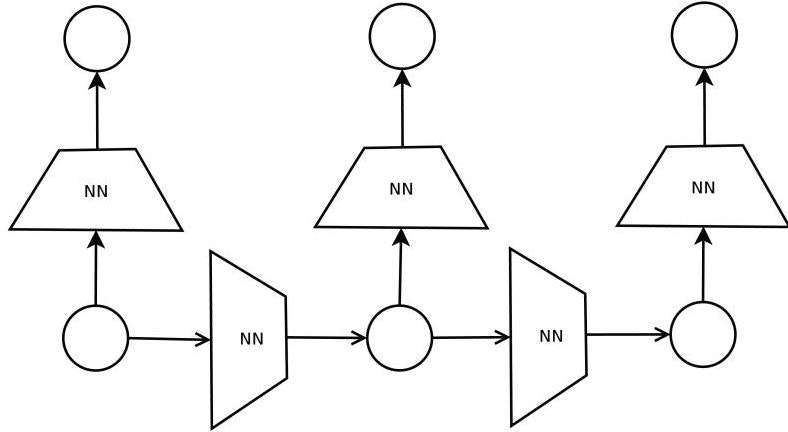
ご存知の方もいると思うが、UGモデルは計算コストがべらぼうに高い。例えば、UGモデルの代表例であるボルツマンマシンでは、モデルのスケールに対して指数関数的にコストが増加する。したがって、計算コストの観点から、入力構造をUGからDAGに落としこむ必要がある。この時使う手法が、アンサンブル学習である。$N$個のノードに対して、各ノードが事後変数になるDAGを(すなわち、$N$種類のDAG)を用意し、全てのDAGをアンサンブル学習にかけるのである。
抽出した特徴$D$は、出力関数にかけられ、アウトプットとして、水の溶解度が計算される。
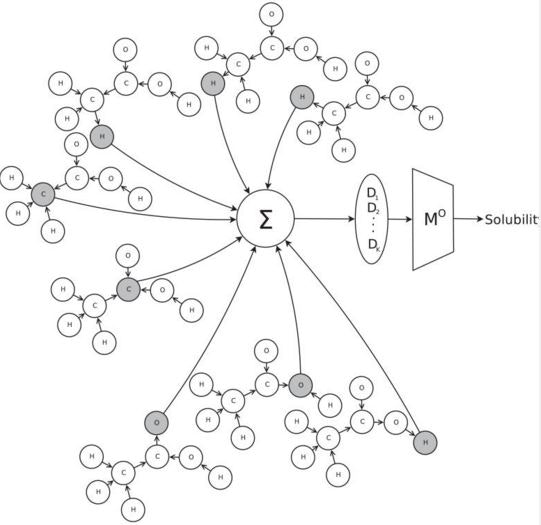
3. モデル適用例
一例として、Huuskonenのデータセットによるテストを挙げる。
DB上の1026種類の分子について、学習した。データは、10グループに分割して、交差確認法にかけている。出力値と教師データとの正答率は、相関係数で表現している。(もちろん、R2=1に近づくほど、正答率が高い)
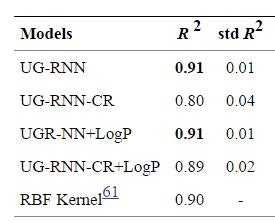
最もパフォーマンスのよかったケースは、RNNに分配係数のデータを付加したパターンだった。