1. はじめに
生命科学データを扱っていると, タンパク質のアミノ酸配列をうまく数値的に表現した分散表現を取得したくなることがあります. 本稿では, タンパク質の1次構造である配列データから深層学習により分散表現を取得する方法を紹介します.
今回の方法では, Word2Vecと呼ばれる自然言語処理(NLP)モデルを配列データのエンコーダ(文字列をベクトルに変換するもの)として扱います[1,2]. 一般的には単語埋め込みと呼ばれるものですが, これをアミノ酸配列に適用します.
モデル自体の扱いは,Gensimというライブラリを用いるとかなり簡易的です. Gensimでは扱えるNLPモデルに限りがありますので, 最新モデルなどは扱えません. しかし, 本稿で紹介するような簡易的な方法でも, タンパク質1次構造を表現した分散表現が獲得できます.
2. アミノ酸配列データのダウンロードと前処理
Protein Data Bank (PDB) を用います. PDBには60万以上のタンパク質アミノ酸配列データが格納されています. そのため, この大規模なデータベースは深層学習モデルの作成に最適なデータベースの1つといえます.
PDBの配列データを利用するには, まず以下のコマンドをターミナルに打ってデータのダウンロードをします.
$ wget https://ftp.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz
ターミナルを使わなくても, PDBのFASTAデータ(配列データ)のダウンロードページから一括でダウンロードできます. 
ダウンロードしたデータを解凍すると以下のようなテキストデータが出てきます.
>101m_A mol:protein length:154 MYOGLOBIN
MVLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRVKHLKTEAEMKASEDLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGNFGADAQGAMNKALELFRKDIAAKYKELGYQG
>102l_A mol:protein length:165 T4 LYSOZYME
MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAAKSELDKAIGRNTNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYKNL
.
.
.
この配列データはアミノ酸配列以外にもDNAの塩基配列なども含まれているため, アミノ酸配列のデータみ抽出して使用します.
f_name = 'pdb_seqres.txt'
f = open(f_name, 'r')
data = f.read()
rows = data.split('\n')
f.close()
protein_sequences = []
flag = False
for r in rows:
if flag==True:
protein_sequences.append(r)
flag = False
if 'mol:protein' in r:
flag = True
print(len(protein_sequences))
3. アミノ酸配列のコーパス化
Word2Vecで自然言語の学習を行うためには, まず文を意味の最小単位に区切って, それを寄せ集めたコーパスと呼ばれるデータセットを用意する必要があります.
具体的にコーパスとはどのようなものでしょうか?
英語を例に考えます.
Newton was inspired to formulate gravitation by watching the fall of an apple.
という文があったとすると, 単純に文字列に変換してWord2Vecに投げるのではなく, 意味の最小単位(英語の場合は単語ごと)に区切る必要があります.
つまり,
'Newton was inspired to formulate gravitation by watching the fall of an apple.'
は, 次のようなデータに加工する必要があります.
['newton', 'was', 'inspired', 'to', 'formulate', 'gravitation', 'by', 'watching', 'the', 'fall', 'of', 'an', 'apple', '.' ]
ここでは, 大文字と小文字を区別しないように表記をどちらか一方に統一する必要がありますが, 小文字に統一するのが一般的です. このような文字列データを大量に寄せ集めたものをコーパスと呼びます. ここまでが自然言語のコーパスを作成する一般的な流れになります.
では, アミノ酸配列のコーパス化について, この自然言語のアナロジーをどのように適用するのでしょうか?
発想は非常に単純で, $n$-gramアミノ酸というものを考えます. $n$-gramアミノ酸とは, 固定された長さ$n$のアミノ酸配列を指します. すなわち,
MVLSEGEWQ...
という配列があった場合の$3$-gramアミノ酸は,
MVL, SEG, EWQ, ...
あるいは,
MVL, VLS, LSE, SEG, EGE, GEW, EWQ...
となります.
このように, 2つの分割パターンが考えられますが, 前者をnon-overlap split, 後者をoverlap splitと呼びます.
$n$-gramアミノ酸を英文における単語, $n$-gramアミノ酸同士の連なりを英文における単語同士の連なりと考えれば, ──英文における単語同士の特定の連結パターンが文法として意味情報を伝えるように──$n$-gramアミノ酸同士の特徴的な配列パターンはアミノ酸配列上の特定の意味情報を伝えるという仮説が考えられます.
このような仮説に基づき, アミノ酸配列のコーパスをWord2Vecに学習させ、特定のタンパク質1次構造の分散表現を獲得するというのが, 本稿の主題です.
では, 実際にアミノ酸配列のコーパスを作っていきましょう. 今回は$3$-gramアミノ酸に基づいたコーパスを作成し, 分割方法は overlap split とします. ※ 分割に結構時間かかります.
from tqdm import tqdm
def n_gram_overlap_split(seq, n):
n_gram = []
for i in range(len(seq)+1):
if i > n-1:
n_gram.append(seq[i-n:i])
return n_gram
copus = []
for seq in tqdm(protein_sequences):
n_gram = n_gram_overlap_split(seq, 3)
copus.append(n_gram)
print(copus[0])
以下のような出力が得られます.
['MVL', 'VLS', 'LSE', ..., 'YQG']
これでモデルのトレーニングのための準備が整いました.
4. Gensimを用いたWord2Vecの学習
Gensimを用いれば簡単にWord2Vecモデルの学習ができます. 使い方も非常に簡単です.
from gensim.models.word2vec import Word2Vec
dim = 1024
model = Word2Vec(
copus,
vector_size=dim,
min_count=5, # ignores all words with total frequency lower than this
window=5, # maximum distance between the current and predicted word within a sentence
sg=0, # 0: CBOW, 1: Skip-gram
workers=-1, # threads
hs=0, # 1: hierarchical softmax
alpha=0.025, # the initial learning rate
min_alpha=0.0001, # learning rate will linearly drop to min_alpha as training progresses
)
model.save("PDBWord2VecCBOW.model") # modelの保存
これだけです. めちゃくちゃ簡単です. lossのモニターが困難などの難点はありますが...
5. Word2Vecを用いたアミノ酸配列の分散表現取得
所望のタンパク質の分散表現を取得するには, まずタンパク質のアミノ酸配列を$3$-gramアミノ酸に分割し, その個々について先ほど構築したWord2Vecから低次元ベクトルを取得します. 次にそのタンパク質の全ての$3$-gramアミノ酸の低次元ベクトルを平均化するという単純な方法で, タンパク質の1次構造に基づいた分散表現を取得します(図参照).
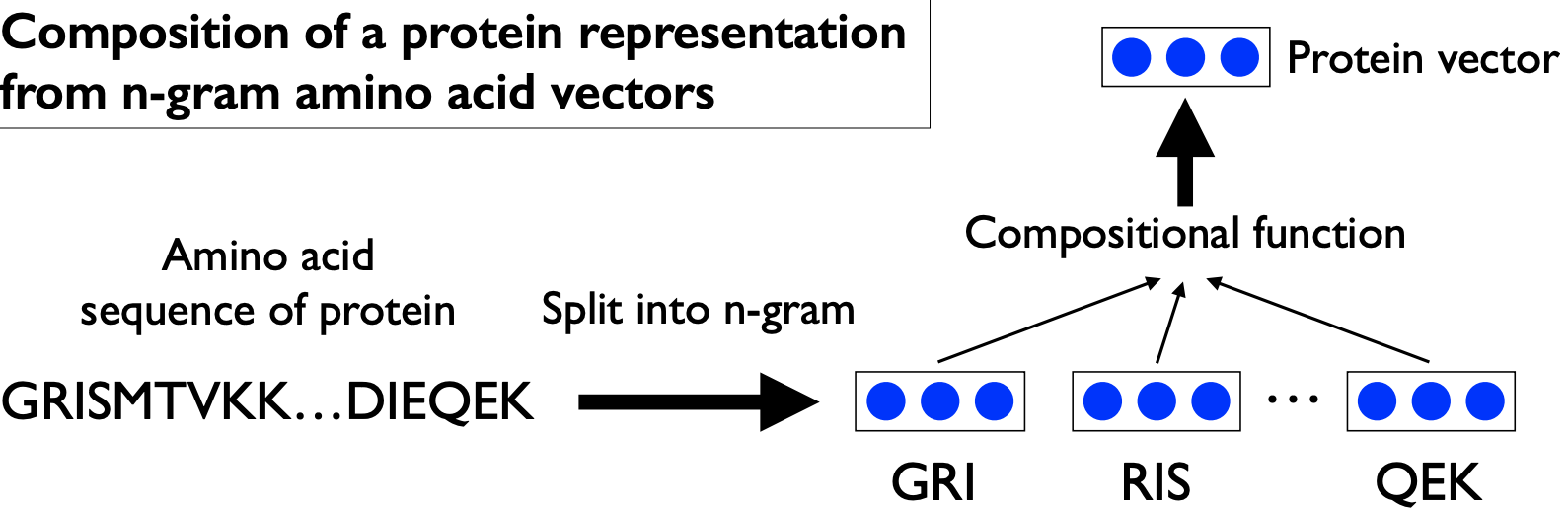
それでは, 早速実装してPDBのタンパク質の分散表現を取得していきます.
先ほど保存した学習済みモデルを読み込み, 計算の軽量化のためにサンプル量を減らして解析していきます.
import random
model = Word2Vec.load("PDBWord2VecCBOW.model") # モデルの読み込み
n_samples = 60000 # 少量の配列データをサンプルにコーパスを作成する
random_state = 1222
random.seed(random_state)
copus_ = random.choices(copus, k=n_samples)
global_vectors = [] # calculate protein global vectors
idxes = []
for i, n_gram in tqdm(enumerate(copus_), total=len(copus_)):
try:
global_vector = np.mean([model.wv[gram] for gram in n_gram], axis=0)
# ベクトル化可能なもののみを取得(出現頻度が低い単語は無視される)
if global_vector.shape[0] == dim:
global_vectors.append(global_vector)
idxes.append(i)
except Exception as e:
print(i)
copus_ = np.array(copus_)[idxes]
print(len(global_vectors))
これで完了です.
試しに取得したベクトルをUMAPで可視化してみます.
from sklearn.preprocessing import StandardScaler
import matplotlib.pyplot as plt
import umap
scaler = StandardScaler()
X_std = scaler.fit_transform(np.array(global_vectors))
mapper = umap.UMAP(
init='random',
random_state=0,
n_neighbors=100
)
embedding = mapper.fit_transform(X_std)
plt.figure(figsize=(11,11))
plt.scatter(
embedding[:,0],
embedding[:,1],
s=0.1,
alpha=0.5
)
plt.ylim([0,10])
plt.xlim([0,10])
plt.show()

6. 配列の類似性を解析してみる
せっかくアミノ酸配列をベクトル化して可視化することができましたので, アミノ酸配列の類似性をUMAPの図に表示してみます.
まず配列の類似性を解析します.
from Bio import pairwise2
def n_gram_decoder(n_gram, n):
amino_acids = []
for i, gram in enumerate(n_gram):
if i == len(n_gram)-1:
amino_acids.append(gram[-(n-1):])
else:
amino_acids.append(gram[0])
return ''.join(amino_acids)
'''
試しにコーパスのインデックス100に該当するn-gramアミノ酸をサンプルとして配列類似性を算出
デコーダーでn-gramアミノ酸からアミノ酸配列に直す
'''
sample_seq = n_gram_decoder(copus_[100], 3)
scores = np.zeros(len(copus_))
for i, n_gram in tqdm(enumerate(copus_), total=len(copus_)):
seq = n_gram_decoder(n_gram, 3)
score = pairwise2.align.globalxx(sample_seq, seq, score_only=True)
scores[i] = score
類似性スコアの分布を見てみます.
plt.hist(scores)

類似性スコア120以上は120を最大値として丸めて, UMAPの図に類似性スコアを色付けします.
plt.figure(figsize=(13,11))
plt.scatter(
embedding[:,0],
embedding[:,1],
s=1,
alpha=0.6,
c=scores,
cmap=plt.cm.viridis_r,
vmax=120
)
plt.colorbar()
'''
index100のサンプルは少し大きめに強調しておきます
'''
plt.scatter(
embedding[100,0],
embedding[100,1],
s=150,
alpha=0.6,
c=plt.cm.viridis_r.colors[-1]
)
plt.ylim([0,10])
plt.xlim([0,10])
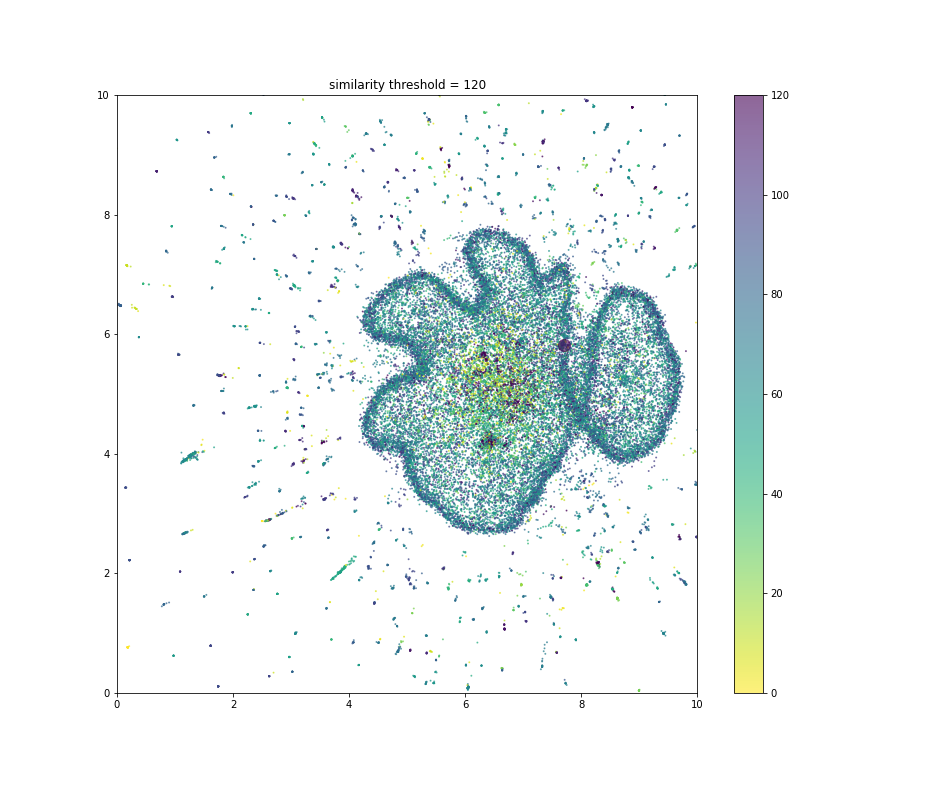
枠みたいに連なっている部分は中程度の類似性, 枠内は類似性が低いことがわかりますね.
また、枠内でも特に類似性の高いクラスタがちらほらみえました.
類似性スコアの最大値を動的に変えたgifをみると多少わかりやすいです.

参考文献
[1] タンパク質構造予測のための表現学習 (椿真史, 新保仁, 松本裕治. 2015, 研究報告バイオ情報学)
[2] Masashi Tsubaki, Kentaro Tomii, and Jun Sese. (2018) Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics. 35(2) 309-318