1. SBVSってなに?
Structure-based virtual screeningの頭文字をとって(SBVS). Structure-based drug design (SBDD)の1分野で, 受容体タンパク質(薬の標的タンパク質)側の構造情報を元に親和性の高い薬をスクリーニングする手法です.
標的タンパク質を”鍵穴”, 医薬品候補化合物(リガンド)を”鍵”とした鍵と鍵穴問題として例えられる医薬品開発において, 鍵穴の情報であるタンパク質の立体構造を顕に扱うSBDDは創薬研究者にとって有用な数々の情報をもたらすそうです.
SBVSにおいて特に重要な役割を果たす手法は, 計算機によって標的タンパク質とリガンドの立体的な結合構造と結合親和性を予測するドッキング計算です. 例えば新型コロナウイルス(COVID-19)の治療薬候補を絞り込んだ時もこのドッキング計算が用いられました.
ホモロジーモデリング → ドッキング → MD とやって低分子の承認薬 1903 個の中から 2019-nCoV Mpro 酵素に阻害活性のありそうな薬剤を絞り込んだ報告。ドッキングは正直、当たるも八卦 当たらぬも八卦なところもあるが、お手本的な手順を踏んでいて勉強になるのでちょっと見てみよう。 https://t.co/oCDvcjbno3
— 叢雲くすり(創薬ちゃん)💉 ㋻済 (@souyakuchan) January 29, 2020
[タンパク質-リガンドドッキングの現状と課題[1]](https://www.jstage.jst.go.jp/article/cicsj/34/1/34_10/_article/-char/ja)によると, ドッキングは以下のような利点があるそうです. >ドッキング計算によってリード化合物の結合構造を正しく特定することができれば,標的タンパク質に対するリガンドの相互作用機序を*原子レベルで理解*することができる.さらにこれらの立体的な結合構造の情報を利用することによって, *医薬品候補化合物の結合親和性や選択性向上のための合理的な置換基の設計*も可能となる.
タンパク質は多数の分子で構成されているため, その立体構造をコンピュータにより計算しようとすると多くの計算コストを要します. そのため, ドッキング計算には以下のような簡略化のタイプがあるようです.
- Rigid Docking: 鍵と鍵穴モデル(Lock-and-Key Model)に従い, タンパク, リガンド共に剛体として扱う.
- Flexible Docking: リガンド側のみ柔軟性を考慮(タンパク質は剛体, Flexible Ligand Model)
- Flexible Side-Chains Docking: リガンドに加えて結合ポケットのアミノ酸側鎖の自由度も考慮(Paritally Flexible Protein Model)
- Flexible Protein Docking: タンパク、リガンド共に柔軟性を考慮(Induced-Fit Model)
2. スコア関数
ではどうやって最適なドッキング状態の計算を行うのか? ドッキング計算の手法には様々なアプローチが存在しますが, 共通する2つの目的は標的タンパク質に対するリガンドの結合構造と, 標的タンパク質に対するリガンドの結合親和性を高速に予測することです. この目的を達成するために, ドッキングプログラムには高精度なスコア関数と効率的な探査アルゴリズムが必要とされます.
2.1.力場に基づいたスコア関数(force-field-basedscoring function)
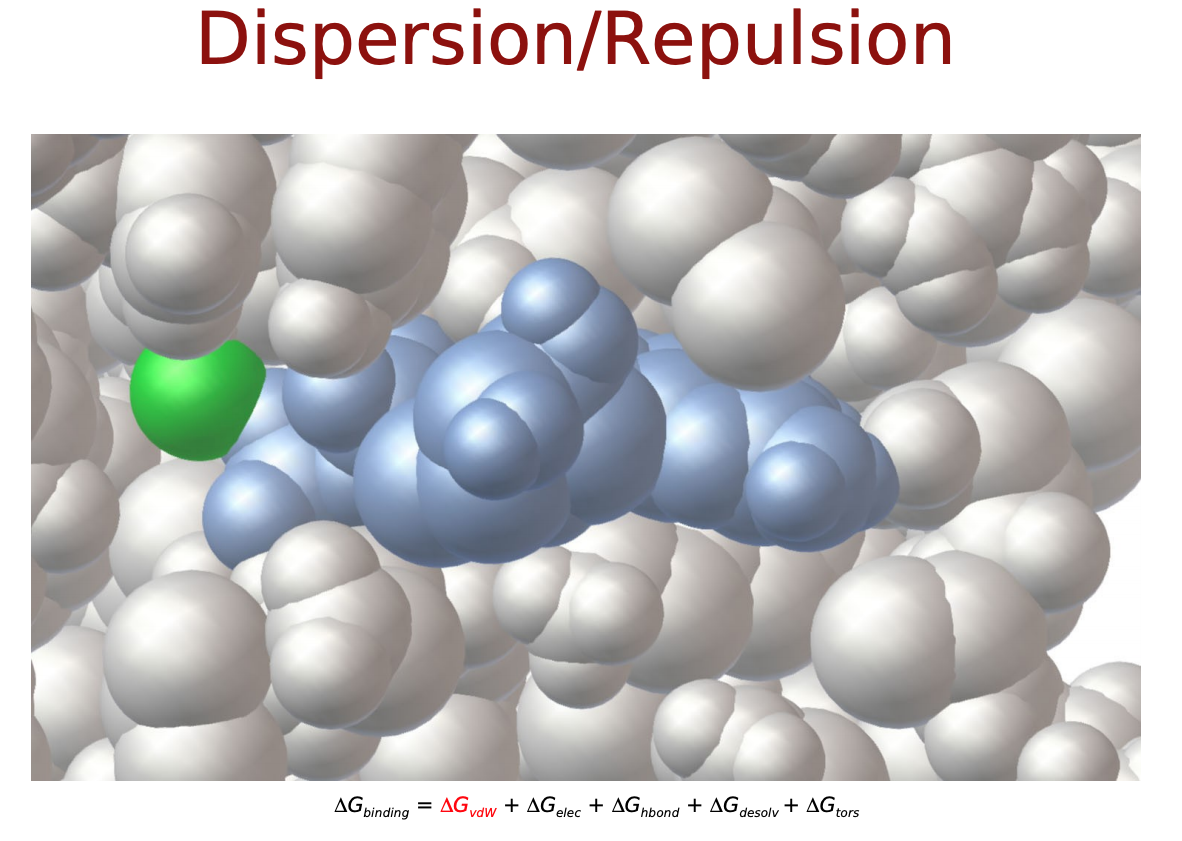
力場に基づいたスコア関数では, タンパク質―リガンド原子間の相互作用はファンデルワールス相互作用 (vdW) や静電相互作用, これらに溶媒効果やリガンドの内部ポテンシャルの変化を記述したものによって表されるそうです. 以下のような物理化学的相互作用を考慮します.
$$
\Delta G = \Delta E_{vdw} + \Delta E_{electrostatic} + \Delta E_{H-bond} + \Delta E_{desolvation} +\Delta S_{conf}
$$
- $\Delta G$:自由エネルギーの変化量
- $\Delta E_{vdw}$: ファンデルワールス相互作用
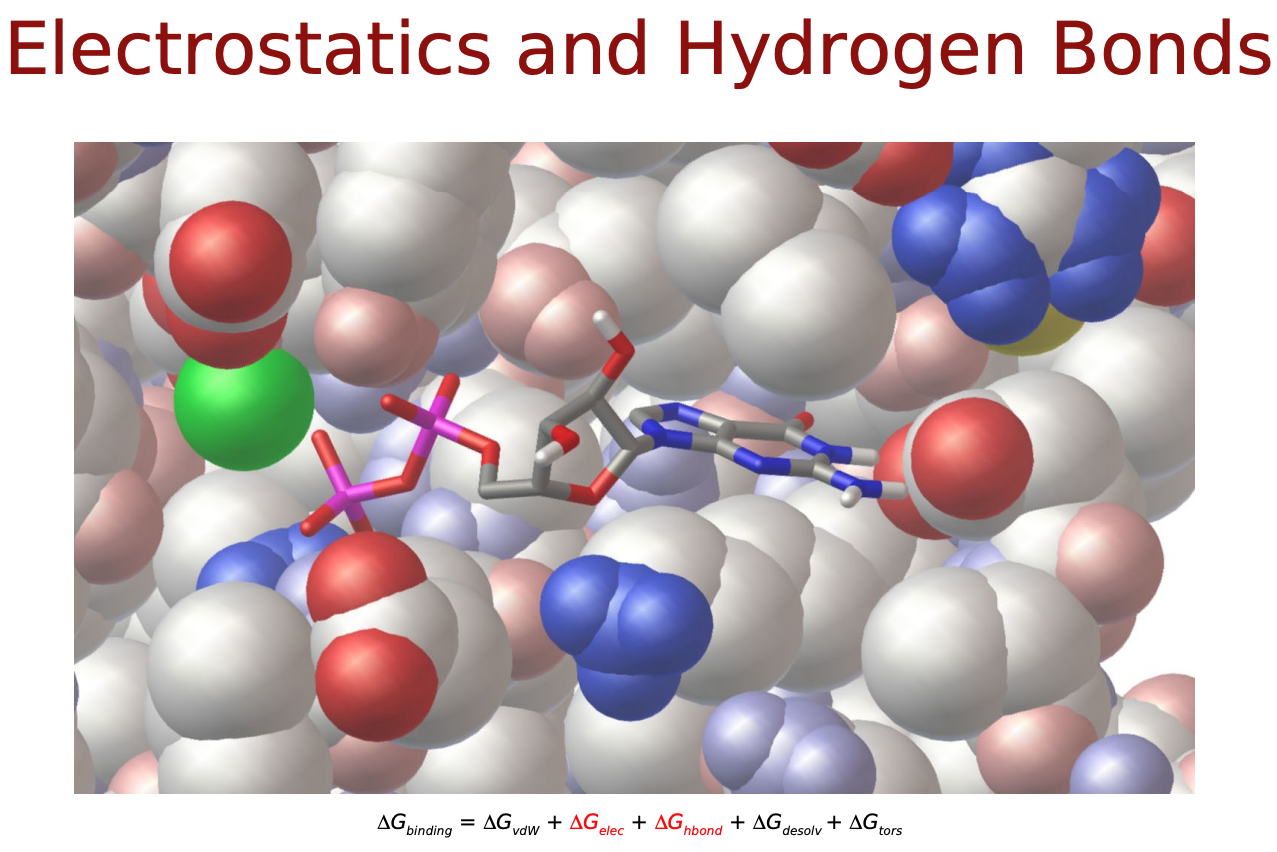
- $\Delta E_{electrostatic}$: 静電相互作用
- $\Delta E_{H-bond}$: 水素結合による相互作用
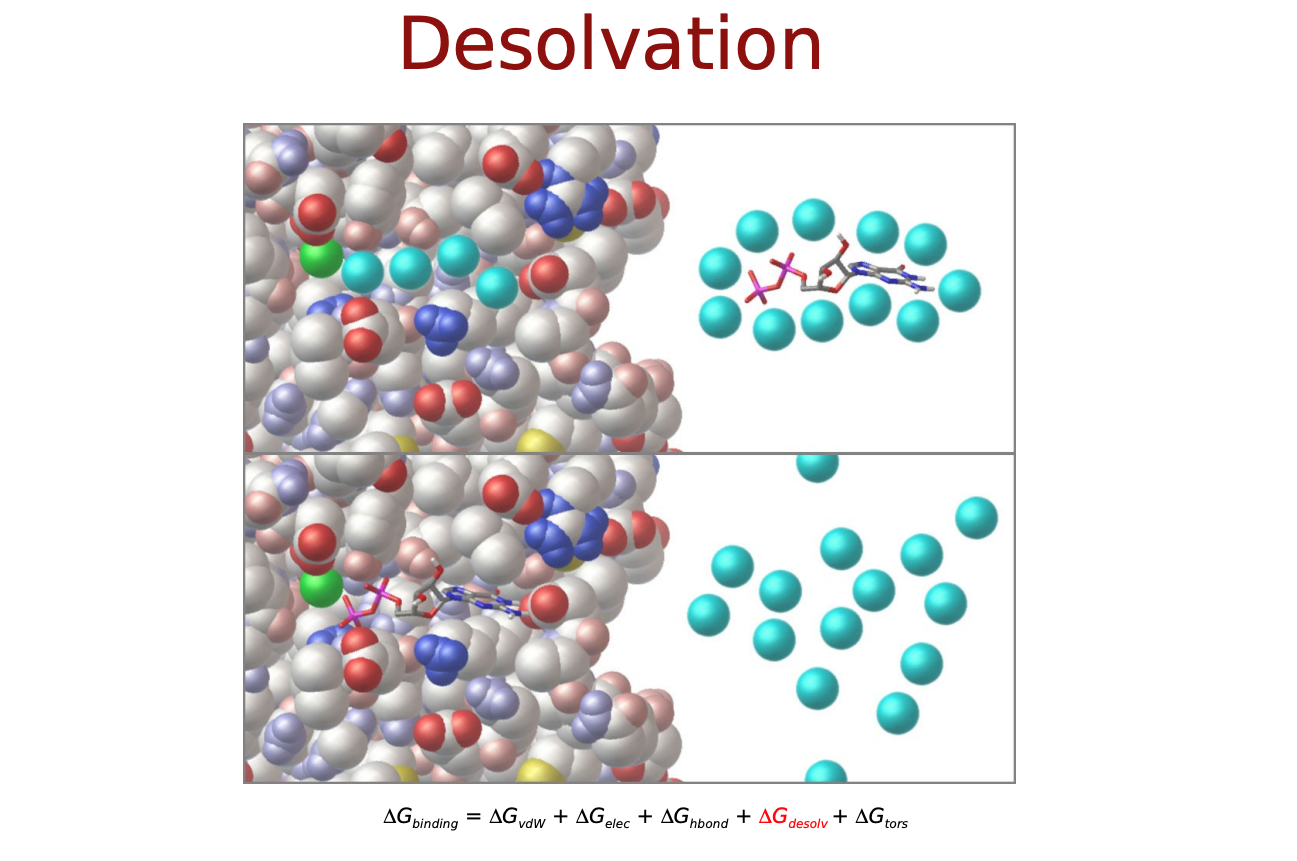
- $\Delta E_{desolvation}$: 脱溶媒効果
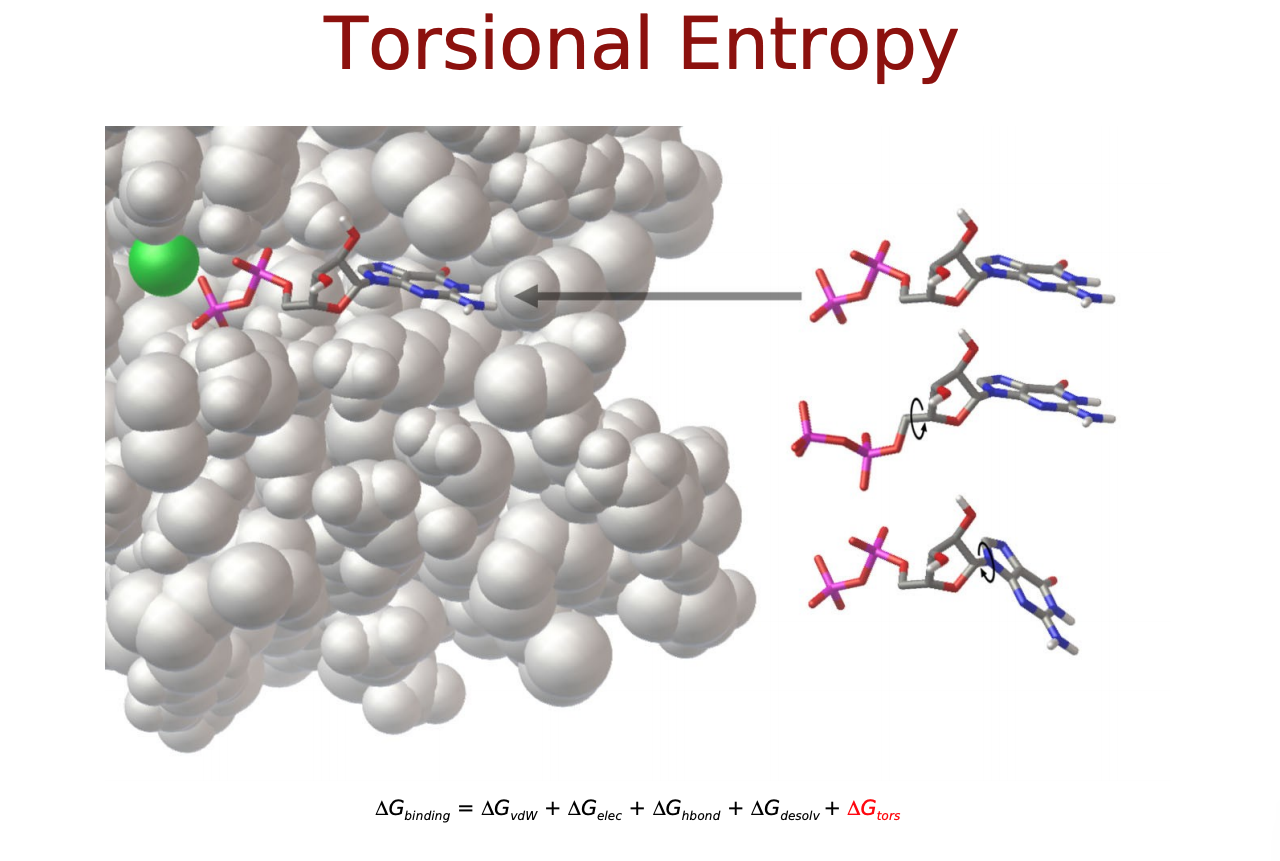
- $\Delta S_{conf}$:リガンドのエントロピー変化の項
$\Delta E_{vdw}, \Delta E_{electrostatic}, \Delta E_{H-bond}$は直感的にも納得できるエネルギー項だと思いますが, $\Delta E_{desolvation}$と$\Delta S_{conf}$はなんでしょうか? リガンドがタンパク質に結合するにあたって, 溶媒(水)に囲まれた環境から, 溶媒が外れ(脱溶媒)タンパク質の結合サイト(より疎水性の高い環境)に移行するという過程をたどると考えます. この時のエネルギー変化を考慮するためのものが$\Delta E_{desolvation}$. また, 溶液中では自由な立体構造(配座、コンフォメーション)をとっているリガンドが, 結合に際してがポケットに合うように固定化されこの自由度が失われます. この分のエントロピー変化を考慮するものが$\Delta S_{conf}$となっているようです.
これらのエネルギー変化量を考え, $\Delta G$が負に大きいとき, それがより安定な構造であると考えられます.
それぞれの項のイメージはWhat is Docking?にわかりやすくまとまっております.
スコア関数は他にも色々あるらしいが詳しくは触れない.
3. Docking Study
3.1. 準備 (Macの場合)
- PyMol: 使えればなんでも問題ありませんが, 自分はGitHubからcloneしてビルド&インストールをした, オープンソースのPyMolを使用しました.インストール方法はGitHubとここに従えばできると思います.
- Chimera: ドッキングの前処理に使います.ここからダウンロードしてインストール
- AutoDock Vina: ドッキングに用います. ここからダウンロード
- SARS-Cov-1のMain proteaseの共結晶構造: このタンパク質(PDB ID: 2GZ7)は2021年現在を世界を大きく騒がせているSARS-Cov-2(通称COVID-19)のMain proteaseと相同性が非常に高い. 右上のボタンからpmdファイルをダウンロード
3.2. ドッキングに向けての前処理 I 【PyMol】
3.2.1. 共結晶構造から標的タンパク質とリガンドを単離する
使うのはPyMolとダウンロードしてきた2gz7.pdbファイル.
前処理のベストプラクティスは正直よく知らない. 今回は共結晶構造から水分子を取り除き, 受容体(標的タンパク質)とリガンドに分割してそれぞれpdbファイルとして保存した.
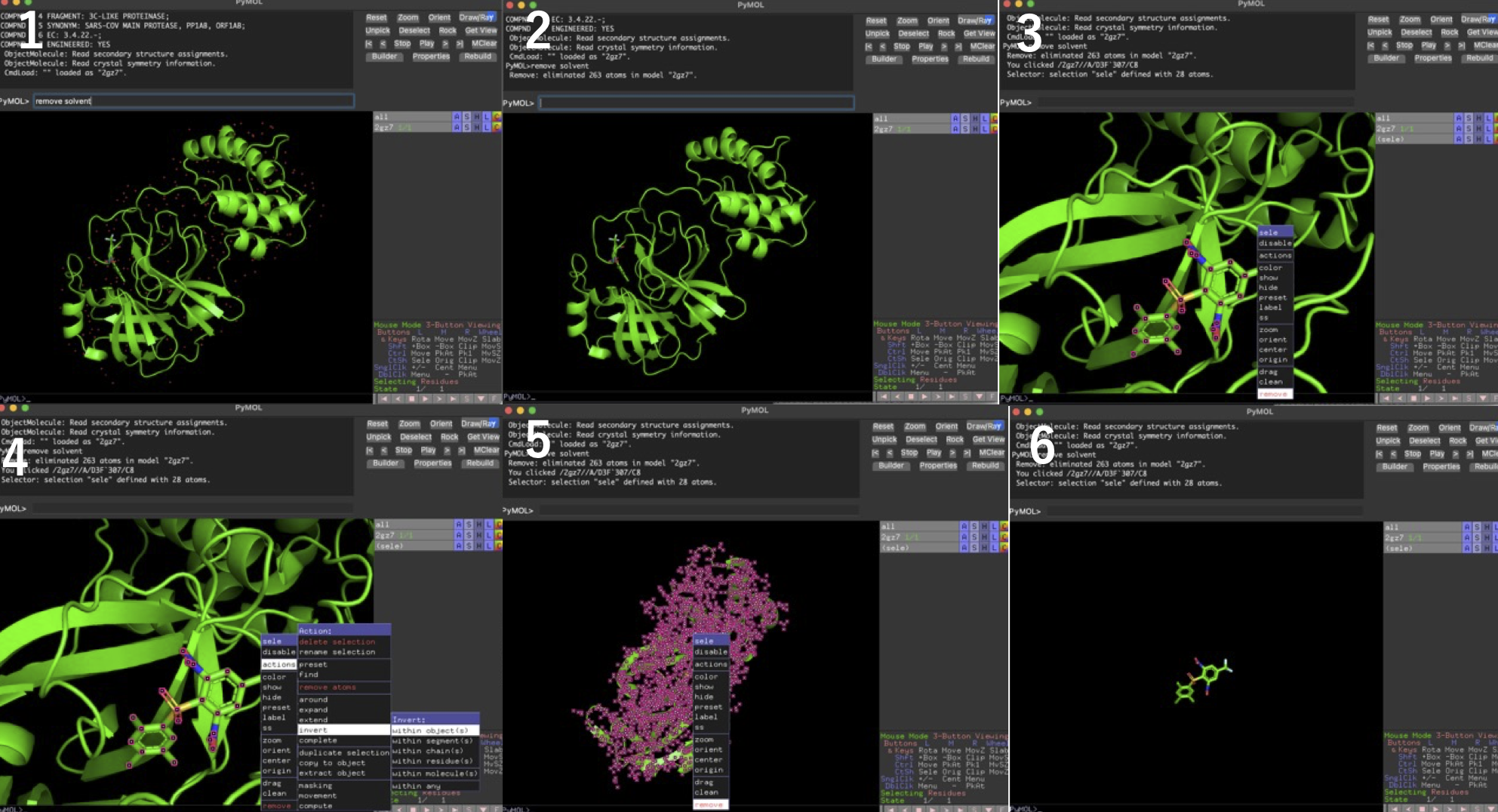
- PyMolのコマンドラインに
remove solventを入力して水を除去 - 水除去後の構造
- リガンドをクリックしてremove→pdbファイルとして受容体を保存
- 「File」→「Reinitialize」→「Everything」してもう一度2gz7.pdbを読み込む. 再度
remove solventで水を除去し, リガンドをクリック→「action」→「invert」→「with object」で選択を反転 - remove
- リガンドをpdbファイルとして保存
3.2.2. ドッキングの中心座標を取得する
共結晶構造のリガンドがはまっていた座標をドッキングの中心座標にしたい.
リガンドのpdbファイルを開く.
リガンドをクリック
以下のコマンドをうつ
pseudoatom center_, sele
xyz = cmd.get_coords('center_', 1)
print xyz
↓実行結果↓
[[-24.638144 -41.232357 5.4246078]]
この座標を覚えておく.
3.3. ドッキングに向けての前処理 II 【Chimera】
単離した受容体とリガンドに水素を付加し, 電荷を割り当てる
このチャプターではChimeraを用います.
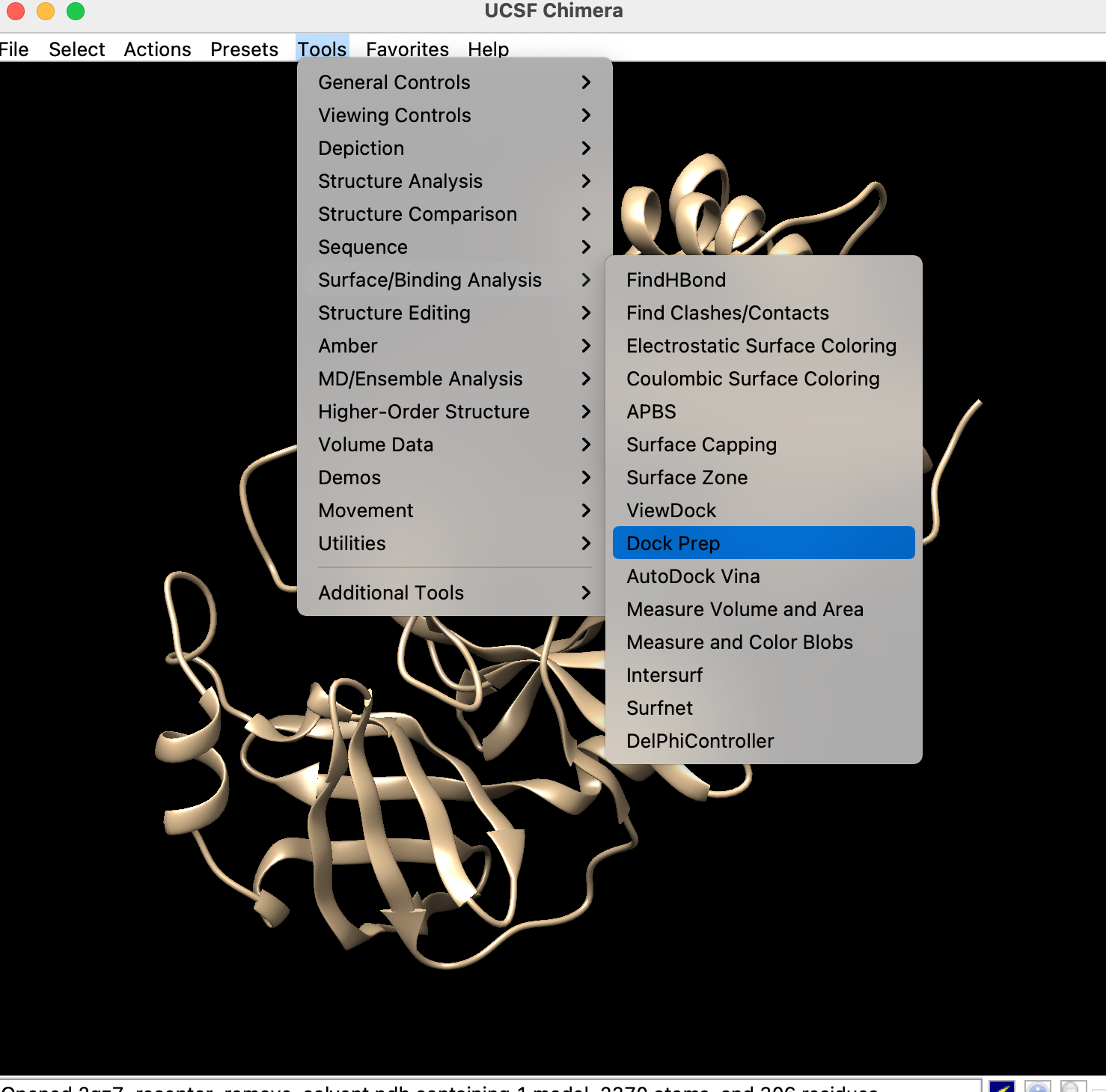
- Chimera起動
- 「File」→「Open」→先ほど作成した受容体のpdbファイルを開く
- 「Tools」→「Dock Prep」
- 「Delete solvent」, 「Add hydrogens」, 「Add charges」 にチェックが入っていることを確認してMol2ファイルに書き出します
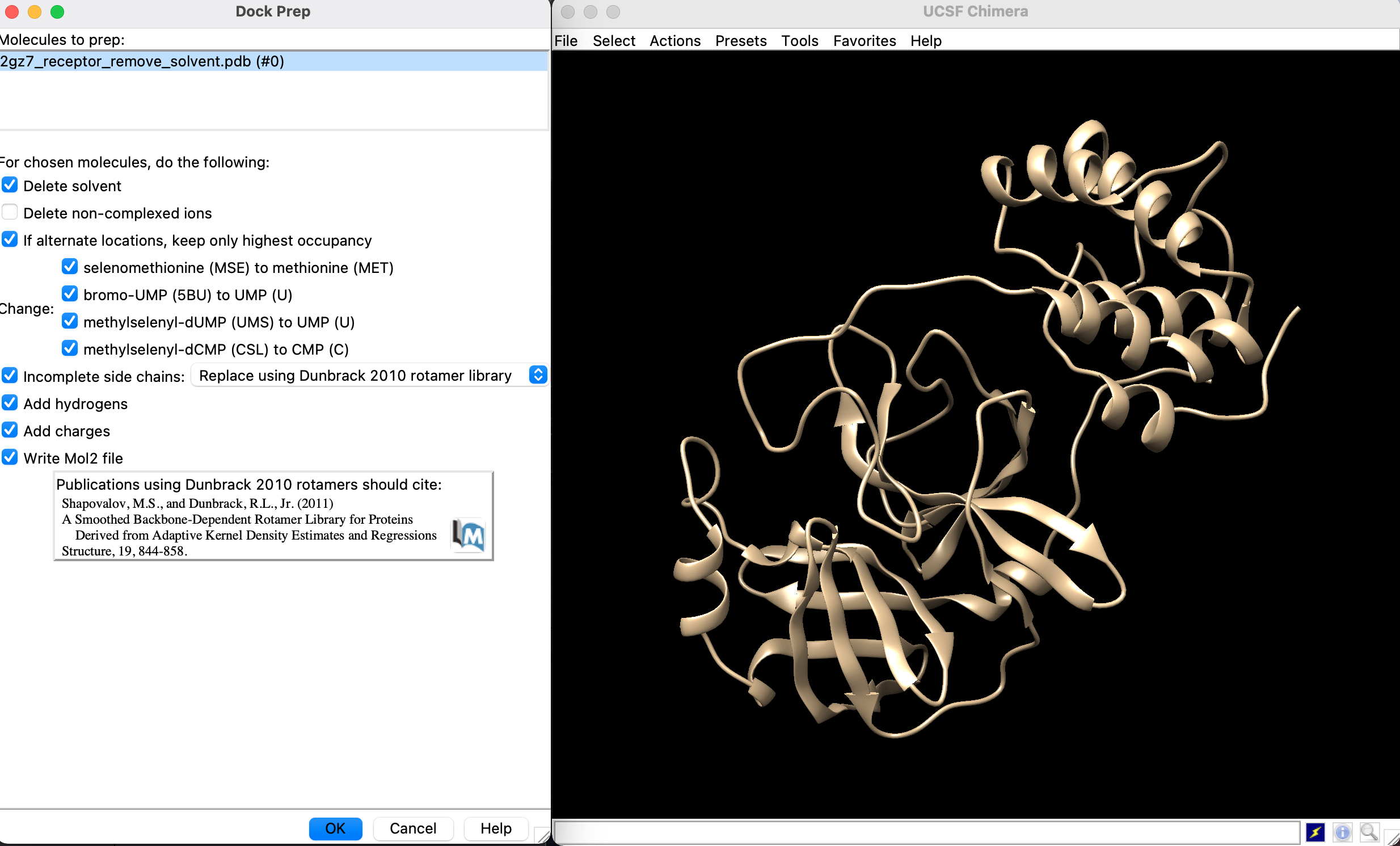
- Add Hydrogens for Dock Prep とかいうウィンドウが出てくるのでOK
- Assign Charges for Dock Prep とかいうウィンドウが出てくるのでOKを押してmol2ファイルを生成します.
- リガンドのpdbファイルに関しても同様に水の除去と水素付加, 電荷の割り当てを行い, mol2ファイルを生成します.
3.4. ドッキングシミュレーション
準備は整いましたのでドッキングを行います.
一旦Chimeraの「File」→「Close Session」によりセッションを終了し, 先ほど前処理した受容体のmol2ファイルとリガンドのmol2ファイルの両方をChimeraで開きます.
以下のような画面になります.
「Tools」→「Surface/Binding Analysis」→「AutoDock Vina」を選択し, 以下の画面のように設定します.
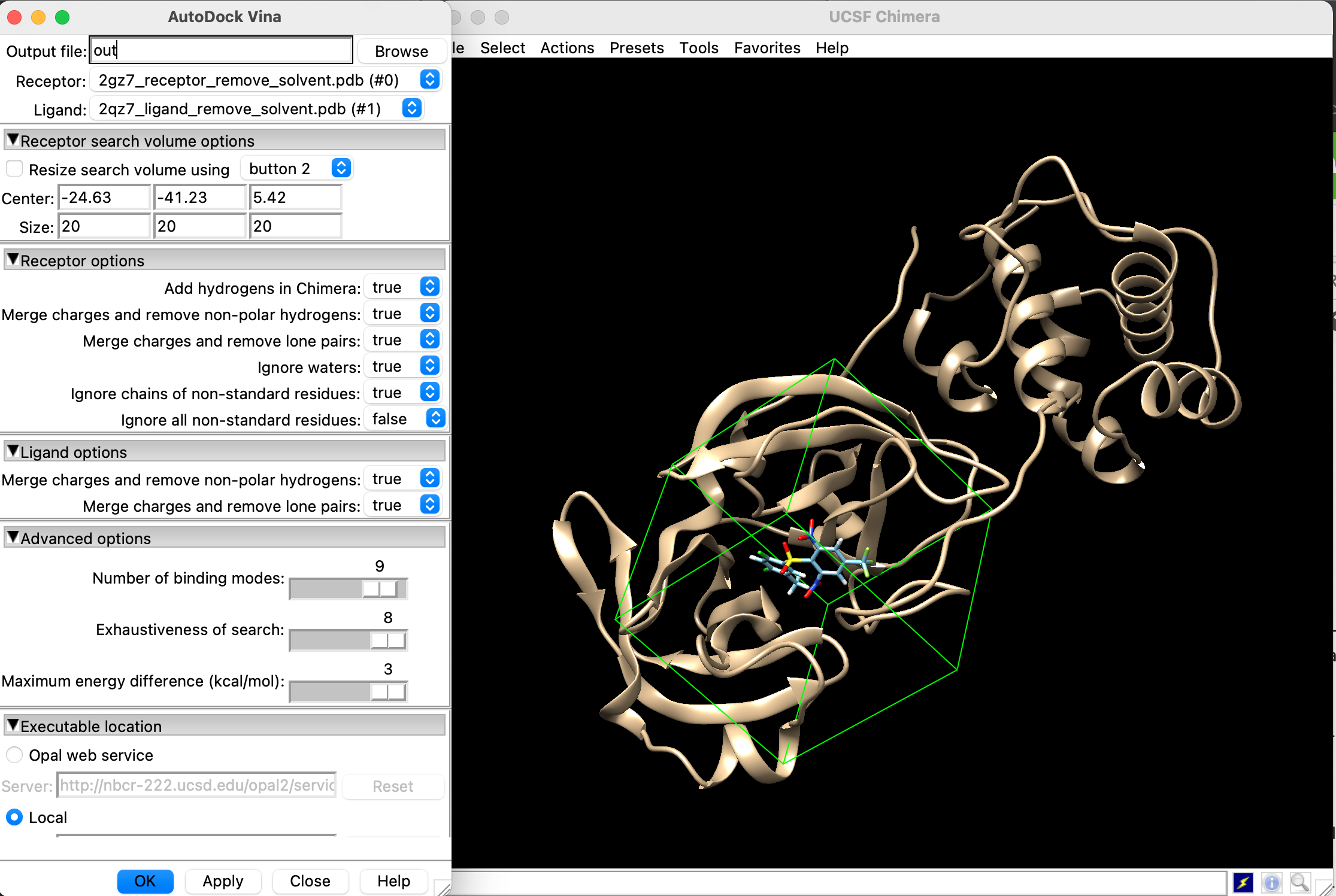
要確認事項
- Receptorには前処理した受容体が選択されているか?
- Ligandには前処理したリガンドが選択されているか?
- Centerには先ほどPyMolで出力した座標が入っているか?
- Localのチェックボックスにチェックを入れ, AutoDock Vina由来の/bin/vinaまでのパスが指定されているか?
- Out fileに出力ファイルの名前が入っているか?
設定が完了したらOKを押すとドッキングが始まり, pdbqtファイルが生成されます
3.5. 結果の解析
3.5.1 結果の解析
画像のような感じで結果が出てきます.
Scoreはエネルギースコアを指しており, これが低い方が好ましい状態です. Root Mean Square Deviation(RMSD)はタンパク質構造の非類似性や誤りの指標として用いられるものです.
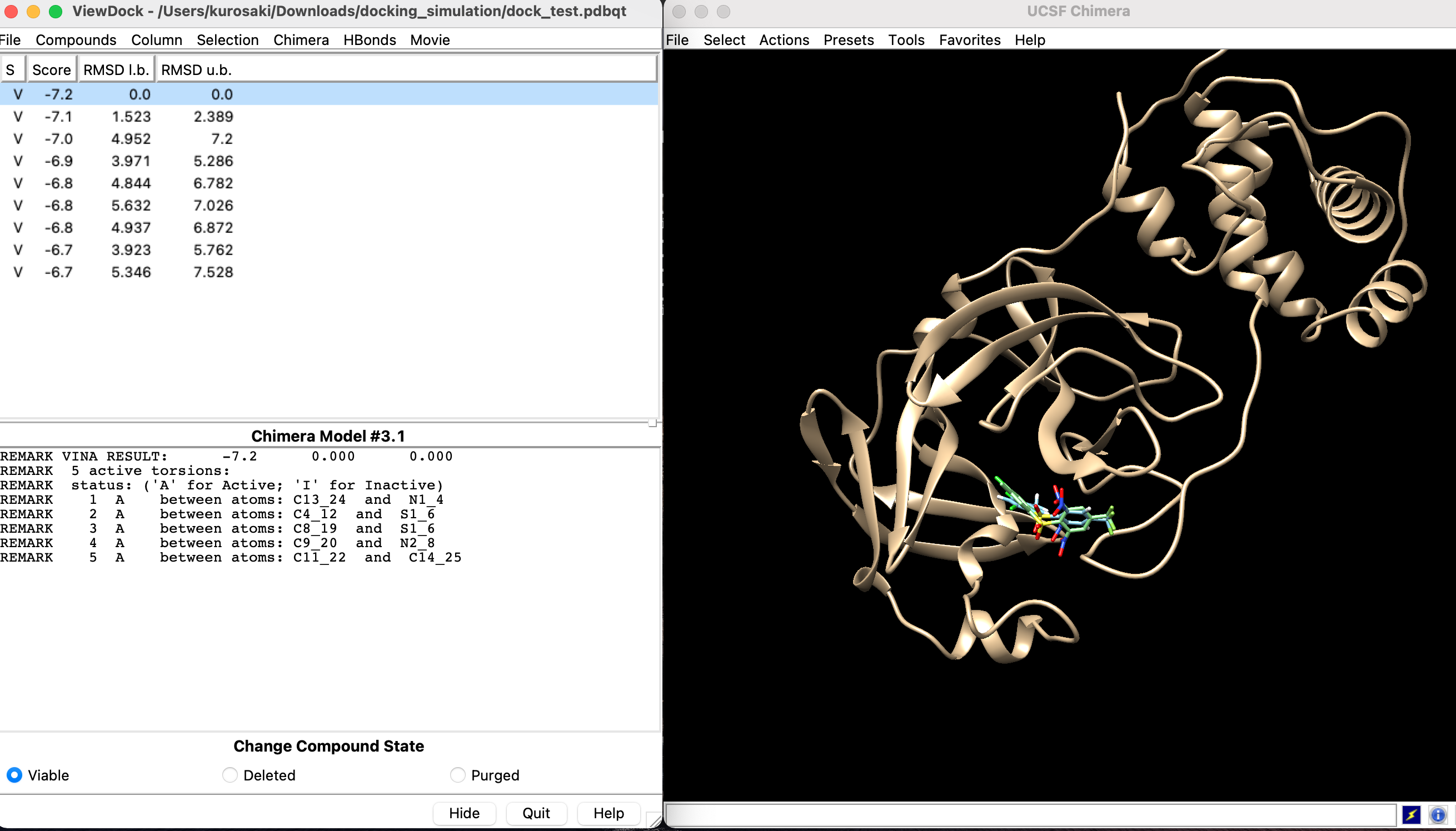
さまざまなドッキングポーズでのエネルギースコアを算出しましたが, スコアが小さい順に並んでいます. この状態で結果を保存します.保存したドッキング結果はChimeraの「Tools」→「Surface/Binding Analysis」→「View Dock」から確認できます.
| ID | Score | RMSD |
|---|---|---|
| 1 | -7.2 | 0.0 |
| 2 | -7.1 | 1.523 |
| 3 | -7.0 | 4.952 |
| 4 | -6.8 | 3.971 |
| 5 | -6.8 | 4.844 |
| 6 | -6.8 | 5.632 |
| 7 | -6.8 | 4.937 |
| 8 | -6.7 | 3.923 |
| 9 | -6.7 | 5.345 |
| という結果でした. |
3.5.2 ドッキングポーズをPyMolで詳しく見る
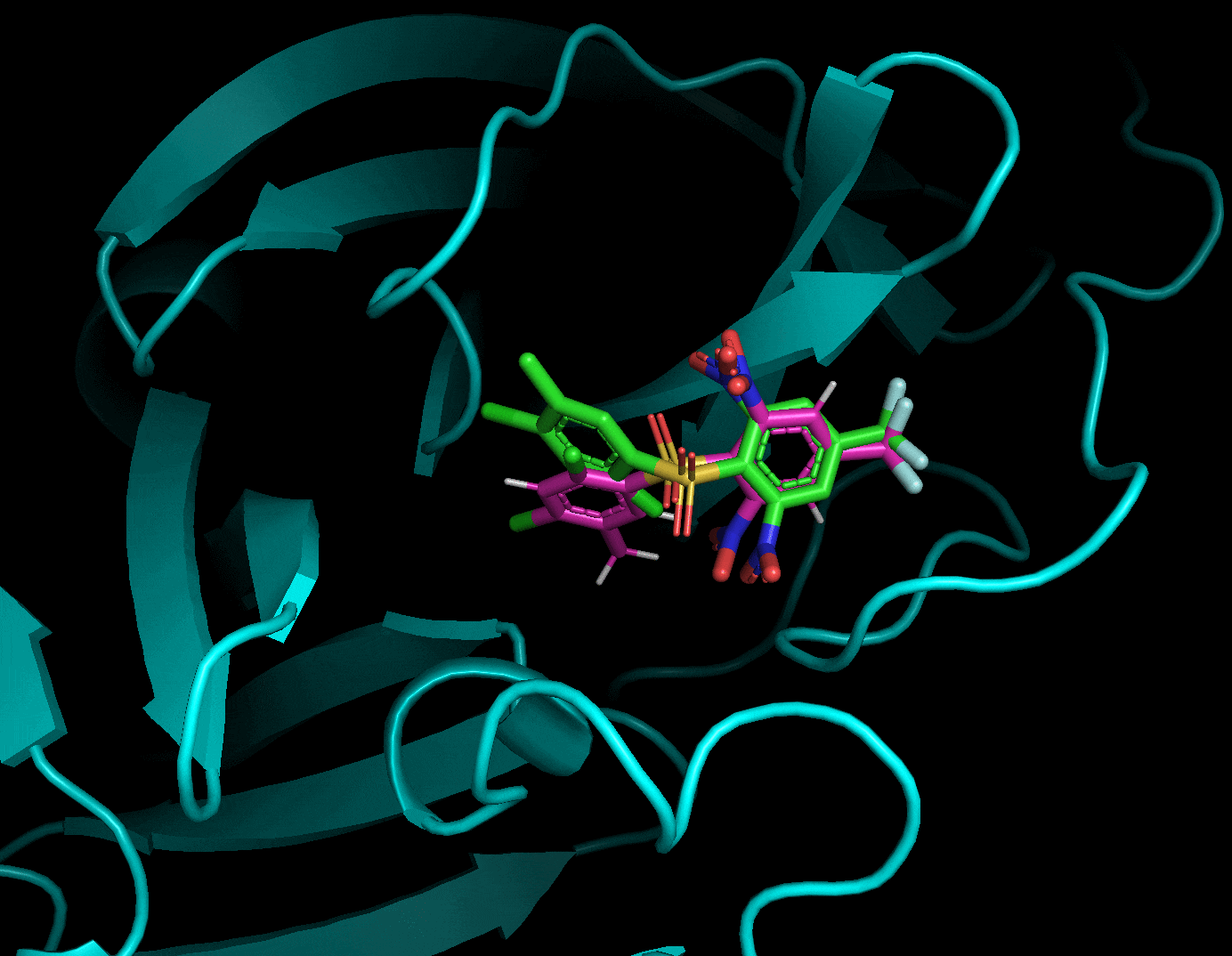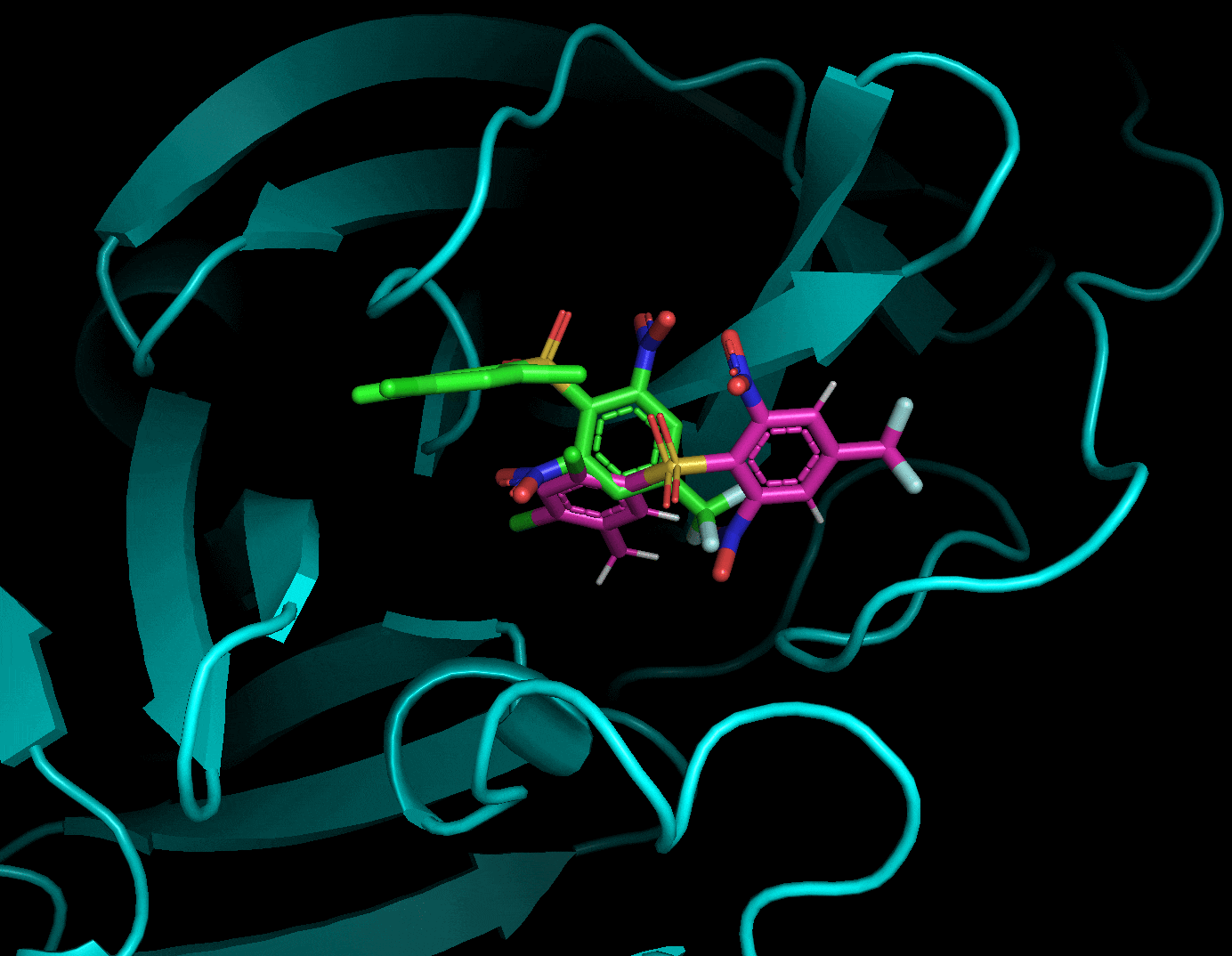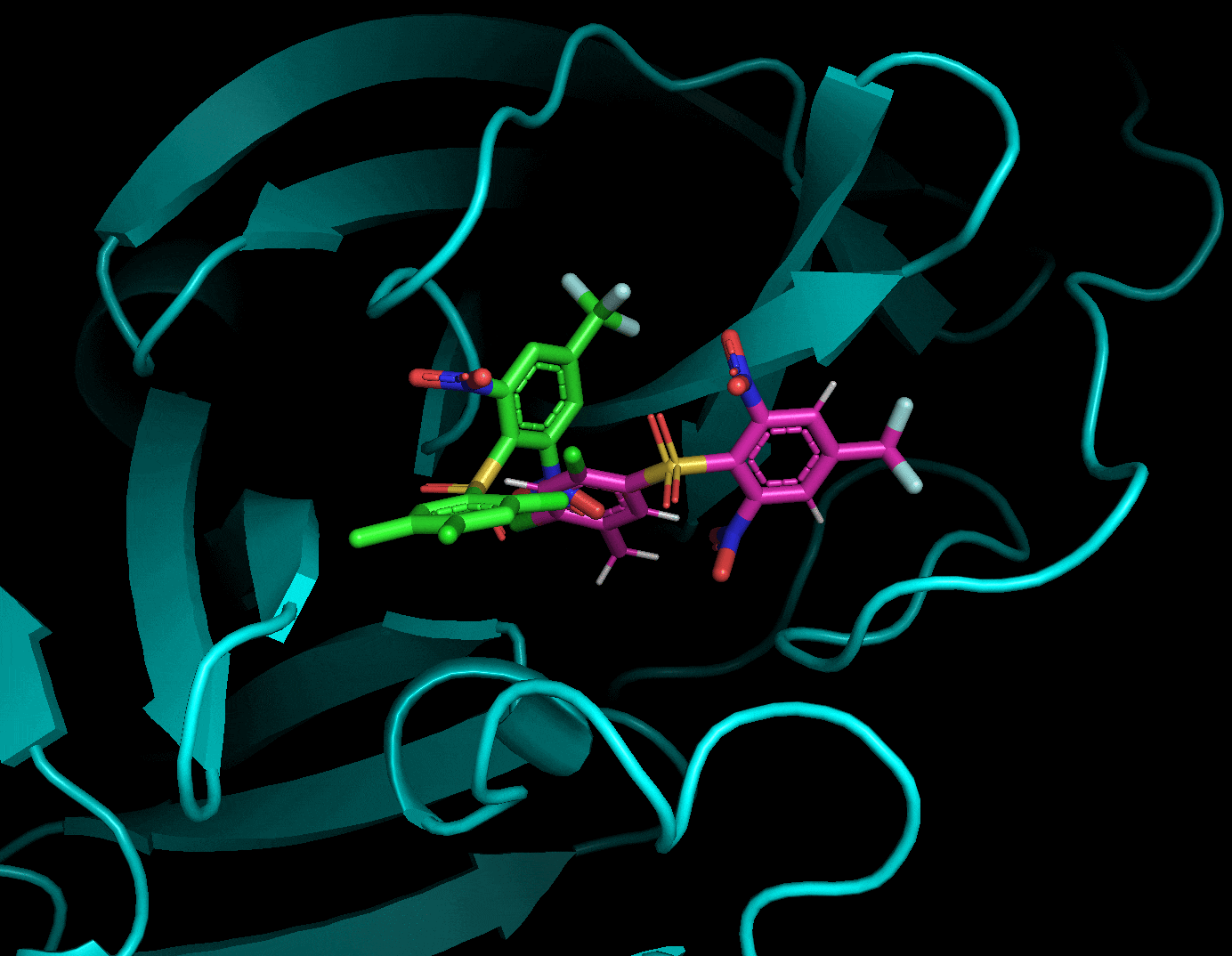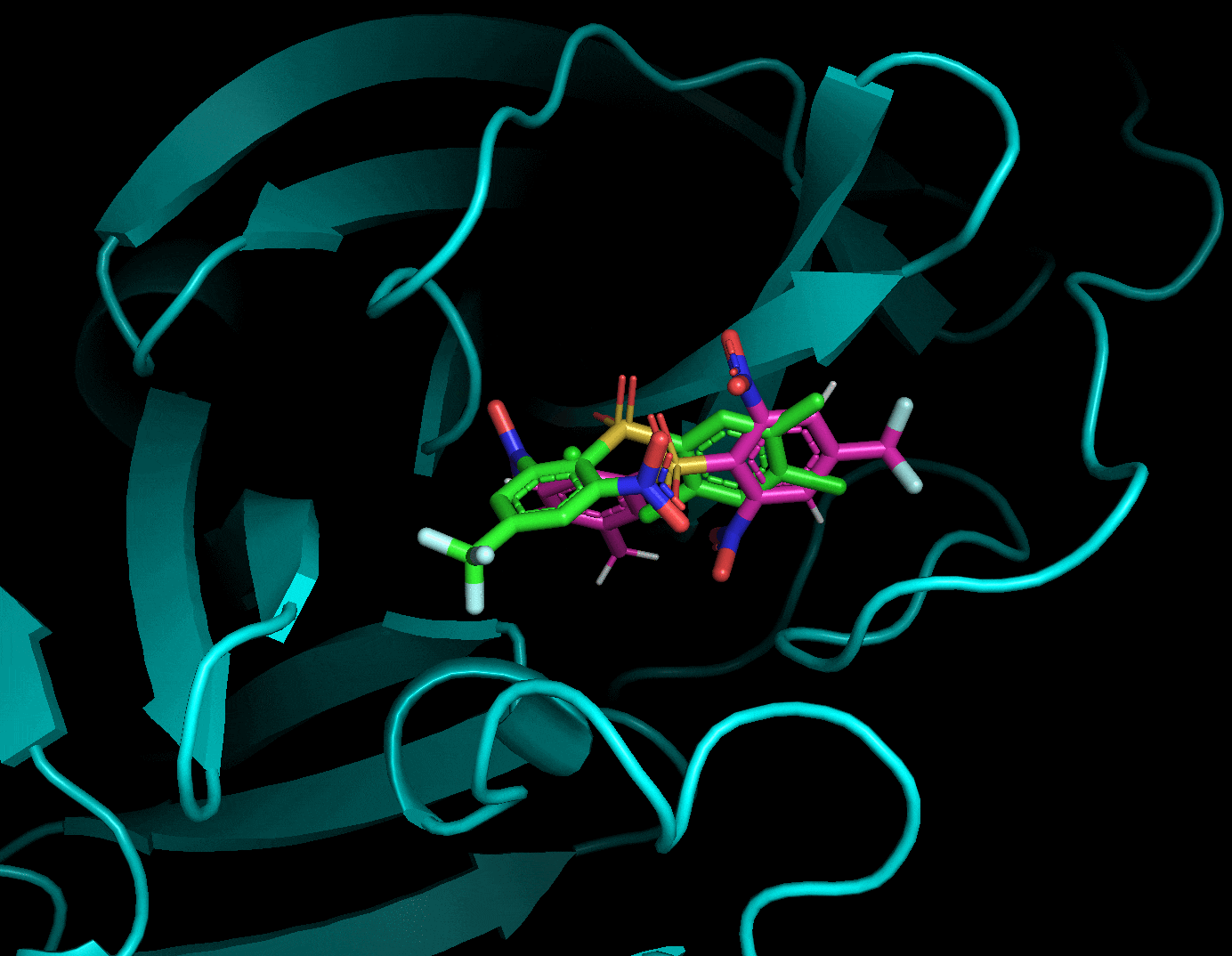
ドッキングした後に生成されたリガンド・受容体・結果のpdbqtファイルを全てPyMolで読み込み, 以下のように確認することができます. 赤がリガンド, 緑がドッキングで計算したドッキングポーズです. 結果のpdbqtファイル(緑)には上の9種のドッキングポーズが格納されています. 右側から「A」→「State」→「split」で全てのドッキングポーズを表示することができます.
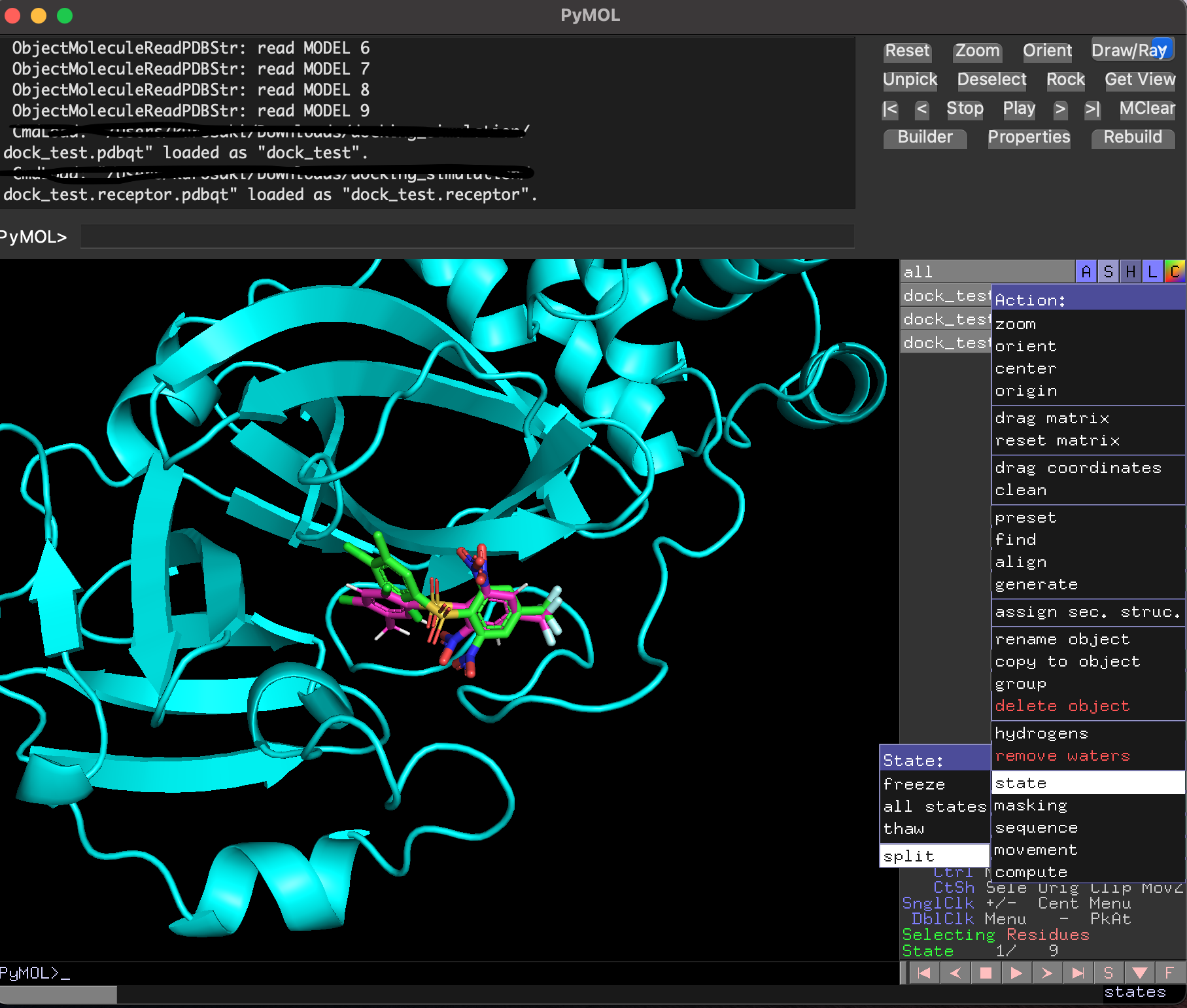
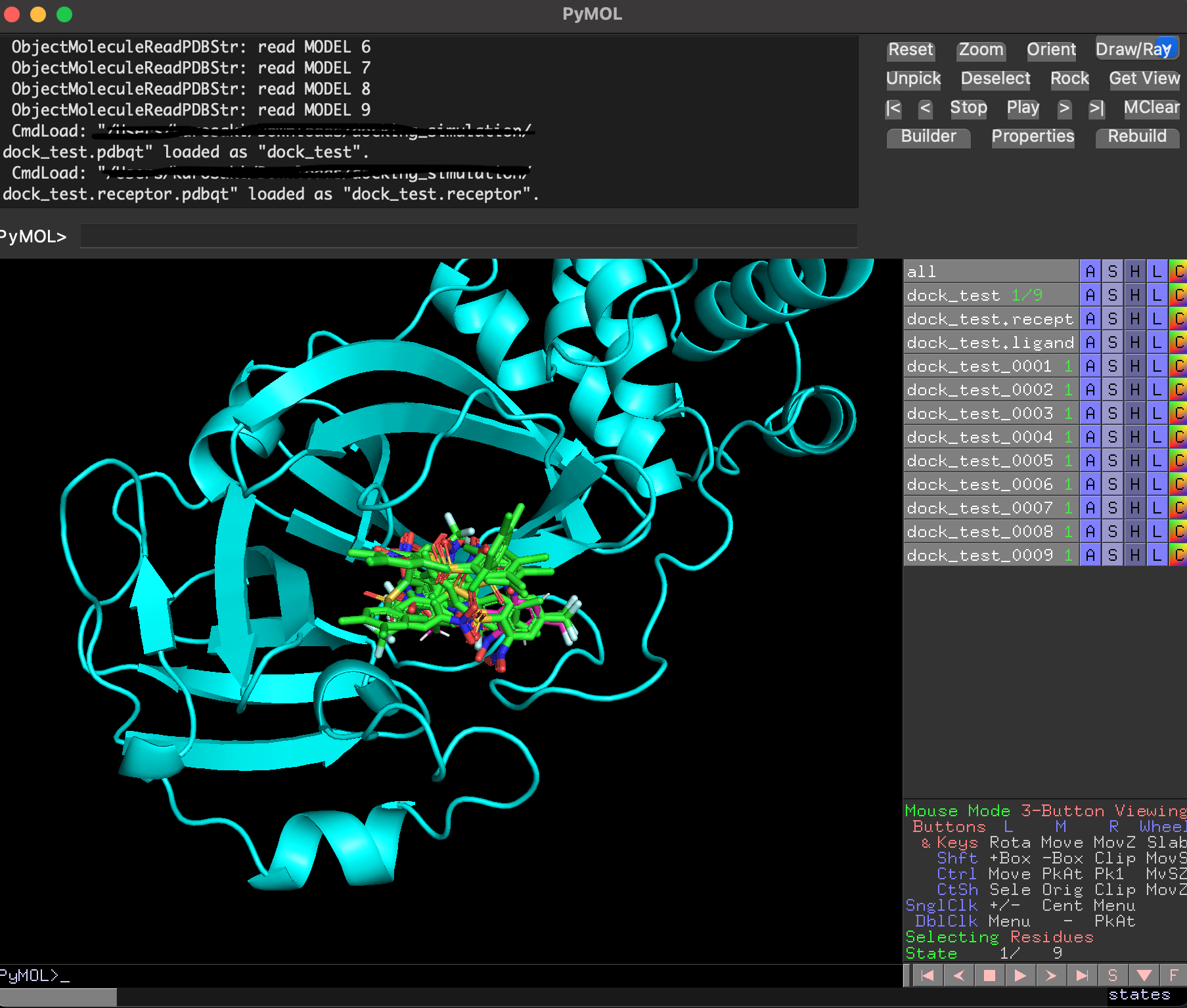
分割したポーズはスコアが低い順に並んでいました.
それぞれのドッキングポーズをgifにしてみました.
3.5. まとめ
SARS-Cov-1のMain proteaseの共結晶構造を用いてドッキングの動作テストを行いました. 使用したソフトは全て無料なので取り掛かりやすいと思います. 今回はドッキングの雰囲気は分かったかなという感触です. しかし実際の活用の面でこのような単純な解析で済むのかという面で複数の疑問が生じました.
- タンパク質の立体構造の最適化は行わないのか?
- 溶媒を除いたが, 生体内ではタンパク質は水に覆われている. 溶媒の影響は考慮しないのか?
- タンパク質のX線結晶構造解析の結果が得られていなかった場合ドッキングは不可能なのか?
この辺に関してまた新たな情報が入ったら記事を作ろうと思います.